
Hi All,
I would like to construct a RangedSummarizedExperiment from an ExpressionSet (eSet) object (microarray data).
I can get an eSet from GEO just fine:
perou2000_gse <- getGEO("GSE61", destdir = inputdatapath, GSEMatrix=TRUE)
perou2000_eset@annotation # returns "GPL180"
and I can get basic annotation information for the array platform:
gse <- getGEO("GPL180").
(There is no bioc annotation pkg for this platform; I used the code listed in here Common workflow to build an microarray annatation package, like hgu133a.db) to test.)
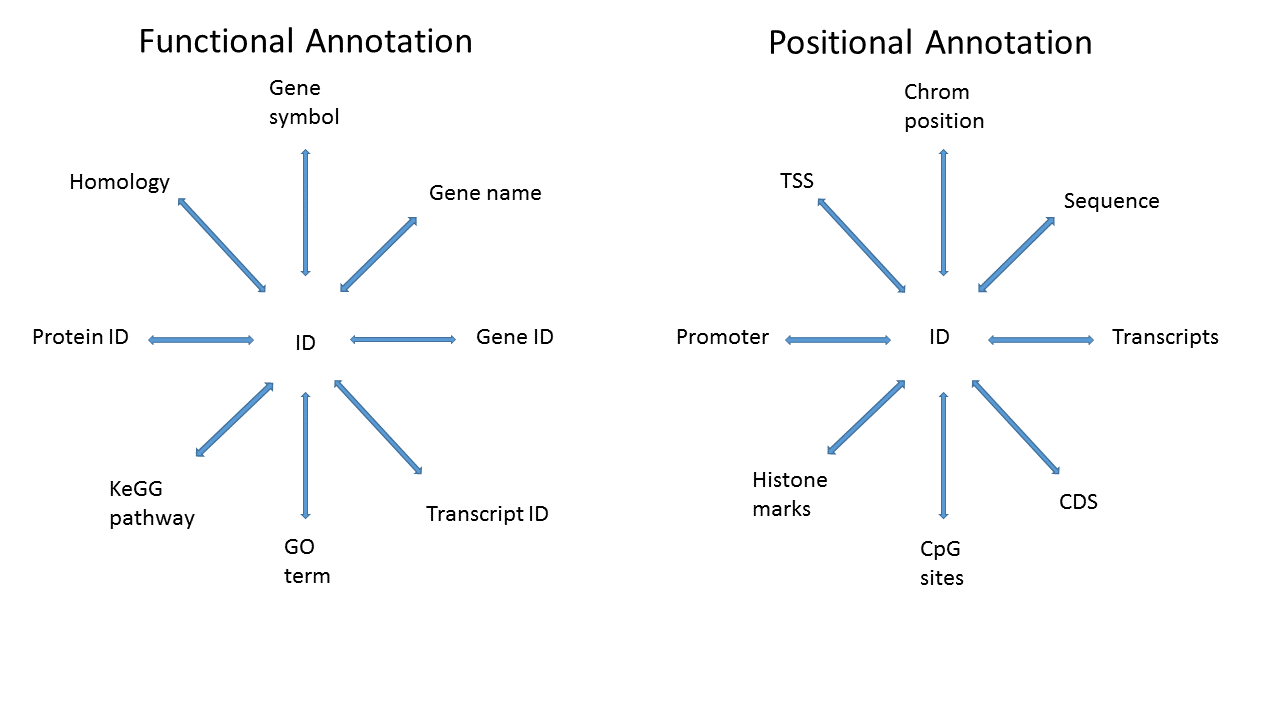
What I would like to do is to construct a RangedSummarizedExperiment object from the eSet. My goal is to have functional annotation (Gene Symbol, GO term, etc..) but also positional annotation (James W. MacDonald | BiocAnno2016) so that I can do range-based queries.
My question is then how to proceed in order to construct an up-to-date and annotation-rich object around this microarray data?
My tentative approach is to take the genbank accession associated with each probe id (info returned by getGEO("GPL180")) and get the respective sequences. Then, somehow, map them to the most recent version of the human genome and work out the rest of the annotation from the found genome locations? Is this how you would approach this problem?
Many thanks in advance

{kind=link}
Thank you for your answer Sean.
Do you see any problem in using this approach:
1. Fetch the EST sequences corresponding to the microarray probe IDs using genbankr pkg?
2. Use gmapR pkg and the human genome BSgenome.Hsapiens.NCBI.GRCh38 to map the sequences.
3. Convert the `GAlignments` objects obtained from `gmapR` to `GRanges` objects and use them to construct the `RangedSummarizedExperiment` object..?
I'd say give it a try.