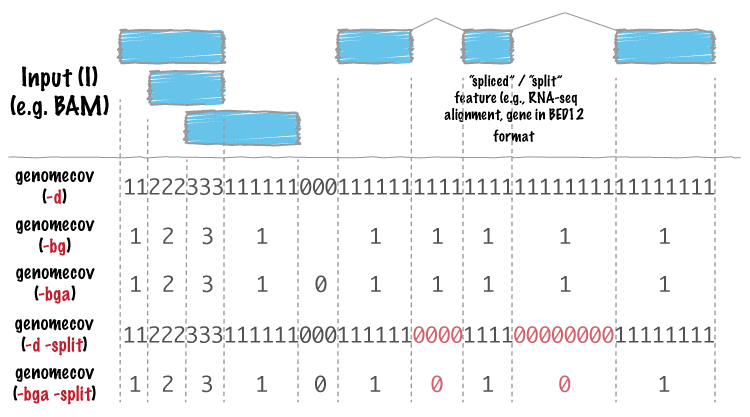
Just to try to give a little bit more context to Martin's suggestion for obtaining the behavior of bedtools -d option. This:
coverage(BamFile("path/to/bam/file.bam"))
is more memory efficient but semantically equivalent to:
gal <- readGAlignments(BamFile("path/to/bam/file.bam"))
coverage(as(gal, "GRangesList"))
As you can see, the GAlignments object returned by readGAlignments() is turned into a GRangesList object before coverage() is called. This coercion preserves the junctions (i.e. the N runs in the CIGAR):
as(GAlignments("A", 20L, "5M10N5M", strand("+")), "GRangesList")
# GRangesList object of length 1:
# [[1]]
# GRanges object with 2 ranges and 0 metadata columns:
# seqnames ranges strand
# <Rle> <IRanges> <Rle>
# [1] A [20, 24] +
# [2] A [35, 39] +
#
# -------
# seqinfo: 1 sequence from an unspecified genome; no seqlengths
However, turning a GAlignments object into a GRanges object does NOT preserve the junctions:
as(GAlignments("A", 20L, "5M10N5M", strand("+")), "GRanges")
# GRanges object with 1 range and 0 metadata columns:
# seqnames ranges strand
# <Rle> <IRanges> <Rle>
# [1] A [20, 39] +
# -------
# seqinfo: 1 sequence from an unspecified genome; no seqlengths
So to get the behavior of bedtools -d option, you could do:
gal <- readGAlignments(BamFile("path/to/bam/file.bam"))
coverage(as(gal, "GRanges"))
or use Martin's memory efficient version above (which should be semantically equivalent).


Hi Jake,
Just to try to give a little bit more context to Martin's suggestion for obtaining the behavior of bedtools
-doption. This:coverage(BamFile("path/to/bam/file.bam"))is more memory efficient but semantically equivalent to:
gal <- readGAlignments(BamFile("path/to/bam/file.bam")) coverage(as(gal, "GRangesList"))As you can see, the GAlignments object returned by
readGAlignments()is turned into a GRangesList object beforecoverage()is called. This coercion preserves the junctions (i.e. the N runs in the CIGAR):as(GAlignments("A", 20L, "5M10N5M", strand("+")), "GRangesList") # GRangesList object of length 1: # [[1]] # GRanges object with 2 ranges and 0 metadata columns: # seqnames ranges strand # <Rle> <IRanges> <Rle> # [1] A [20, 24] + # [2] A [35, 39] + # # ------- # seqinfo: 1 sequence from an unspecified genome; no seqlengthsHowever, turning a GAlignments object into a GRanges object does NOT preserve the junctions:
as(GAlignments("A", 20L, "5M10N5M", strand("+")), "GRanges") # GRanges object with 1 range and 0 metadata columns: # seqnames ranges strand # <Rle> <IRanges> <Rle> # [1] A [20, 39] + # ------- # seqinfo: 1 sequence from an unspecified genome; no seqlengthsSo to get the behavior of bedtools
-doption, you could do:gal <- readGAlignments(BamFile("path/to/bam/file.bam")) coverage(as(gal, "GRanges"))or use Martin's memory efficient version above (which should be semantically equivalent).
H.