
Hi,
I would like to know if it's possible to distinguish between up and down regulated feature after running the stattest function in the ballgown package. In the github page refers to this "For two- and multi-group comparisons, a significant result indicates that the feature is differentially expressed in at least one of the groups", but i'm interested to know in which of the groups the feature is over or under expressed. The displayed fold change values are always positive, so i'm wondering if it's possible to extract this kind of information with this package.
Furthermore, FDR values seem weird, as in all the features with pvalue < 0.05 there isn't any with qvalue < 0.05 too. Why does this happen? Does it has something to do with the statistical model employed in ballgown?
Number of significant features:
> sum(bgresults_c1c3$pval < 0.05, na.rm = TRUE) [1] 2759
Subset with signigicant features:
> subset <- subset(bgresults_c1c3, bgresults_c1c3$pval<0.05, na.rm=TRUE)
Histogram of qvalues:
> hist(subset$qval, main='Ballgown q-values: c1-c3 comparison', col="grey", xlab='Range of q-values for the more significant transcripts')

Thanks in advance,
Pedro Barbosa


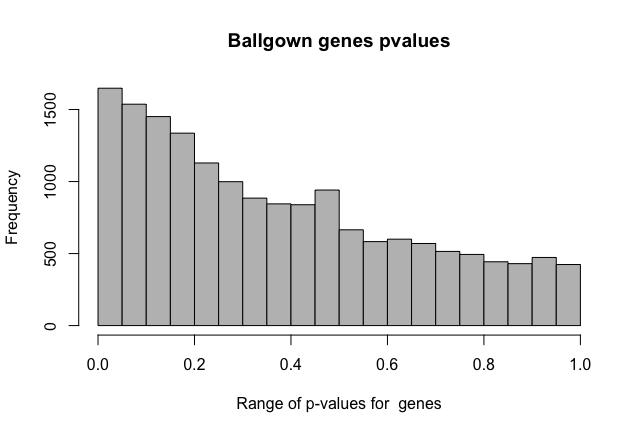
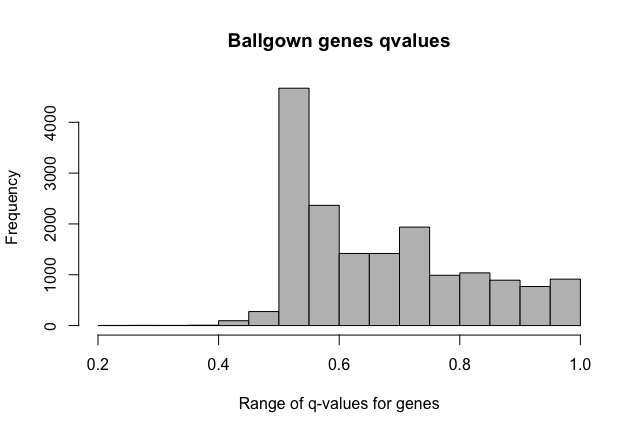
I'm doing two-group comparisons. Thanks about the fold change clarification. I expected fold changes to be outputted as the log2 function (as edgeR, DESeq2) so i would have negative values for the down regulated features.
I did read the paper, but biology is my background, so i've been in trouble to understand what nested llinear models mean in this context. What i observe is when i do multi-group comparisons the range of q-values is more acceptable, but i'm not interested on this type of results. Since the signal might not be strong, should i rely in counting methods rather than estimated abundances ?
Another issue relates to the gene expression calculation. Sometimes i get this following error, am i doing something wrong ?
Hi Alyssa,
I just have started to use Ballgown and R. I have some questions for you:
1) How is it possible to make multiple comparisons (I have 4 different treatments);
2) How does the program identify the replicates?
3) how can I use the ≥tFC in stattest?
Thank you,
Isadora