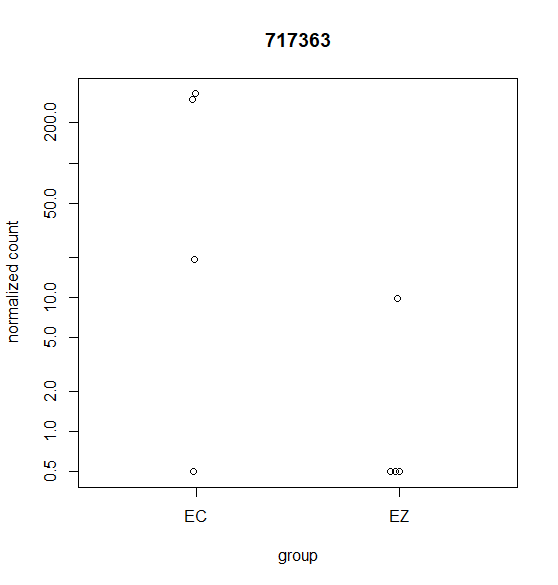
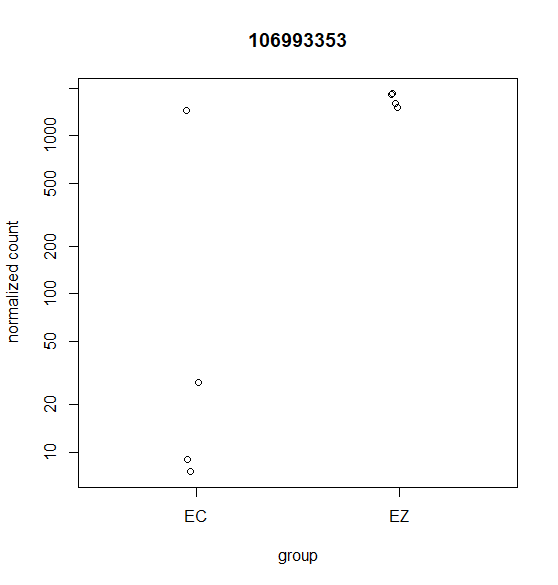
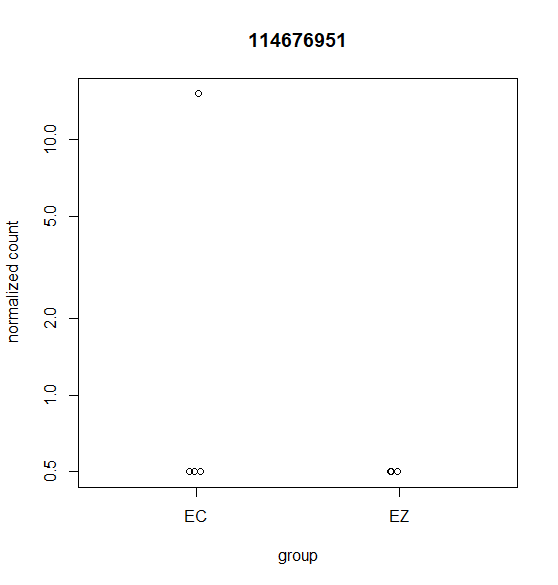
Hi, this is my first time doing differential expression analysis and I have a few questions about the data output. My data was collected from 4 different cell lines derived from 4 different animals (as biological replicates). We are getting inconsistent calling of DEGs and I'm not sure why. Attached are a few scatter plots of the genes in question.
For (B) it was called DEG even though it was only detected in one sample. For (A & C) these were NOT identified as a DEG with DESeq2 but were identified with edgeR's glmQLfit().
For DESeq2 I ran this with the default parameters. Any suggestions on which parameters I could change to account for variation between replicates? Are outliers handled differently when 3 out of 4 samples are highly expressed compared to when only 1 out for 4 samples are highly expressed?
I can add the code if that would help but it's more of a question about the algorithm itself. I can also provide more examples if that would be helpful. Thank you!