
I am reanalyzing some CRISPR-Cas9 screening data to look for sgRNAs effective across cell lines.
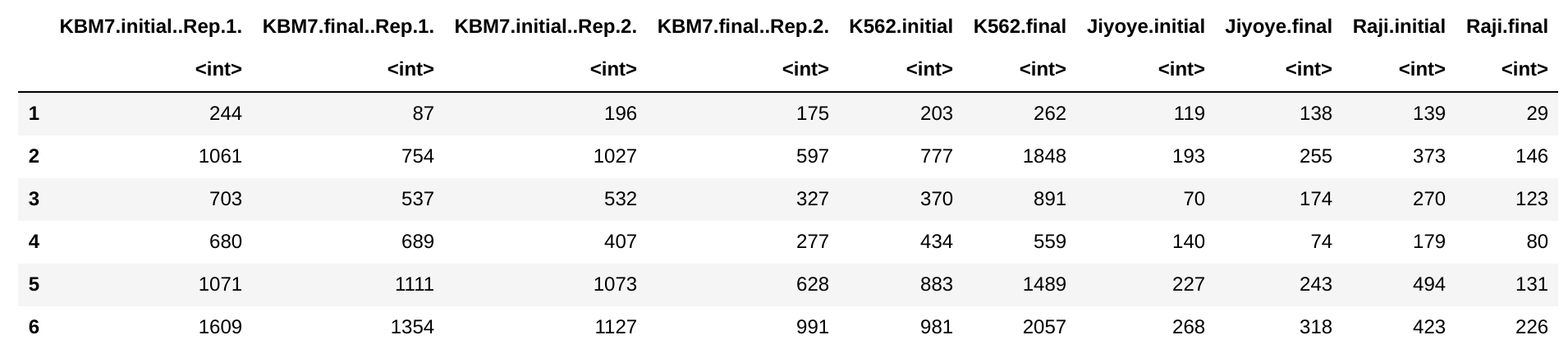
countData:
colData:
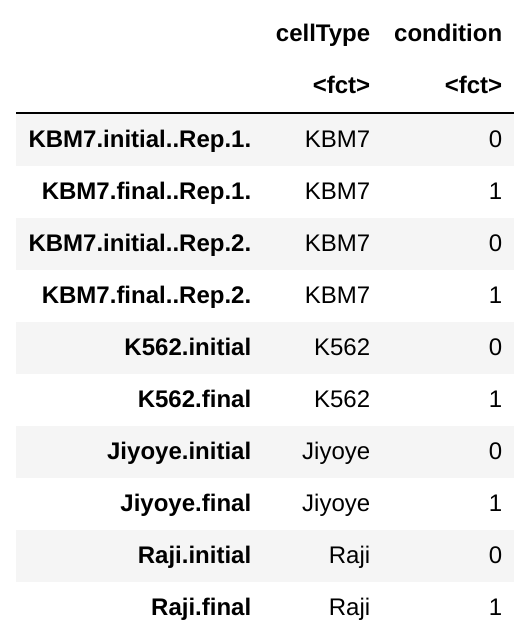
Condition here corresponds to time (initial vs. final).
I can set up the DESeqData like this, where the fold-change result is from the effect of time:
DESeq2::DESeqDataSetFromMatrix(countData = counts, colData = colData, design = ~ condition)
Or I can try to correct for the fold-change that results from cell line:
DESeq2::DESeqDataSetFromMatrix(countData = counts, colData = colData, design = ~ cellType + condition)
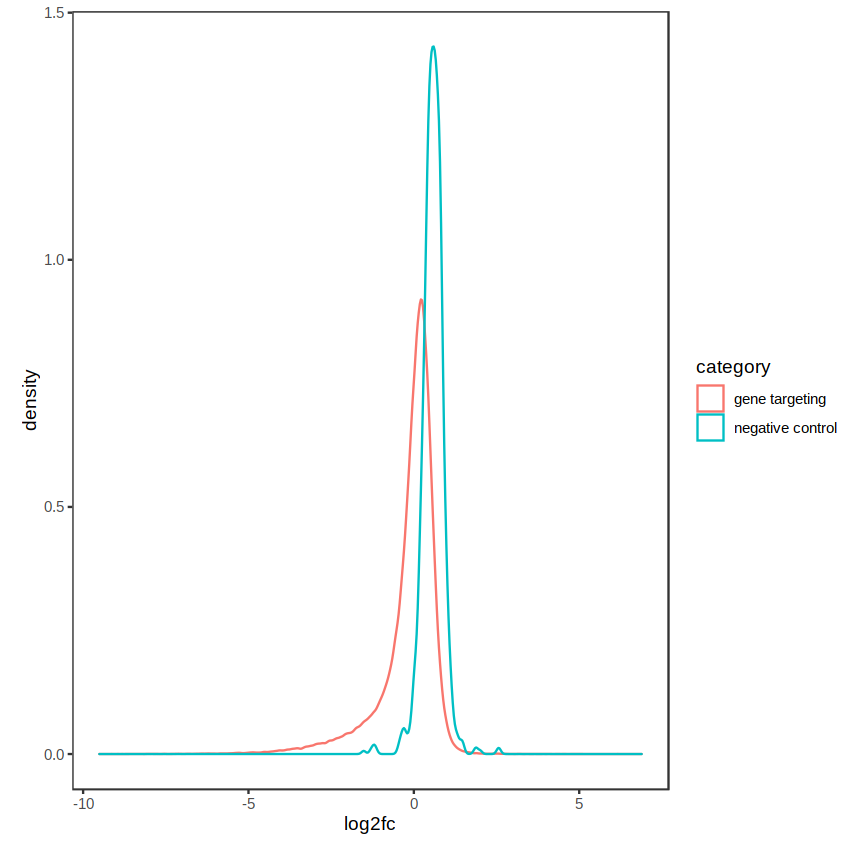
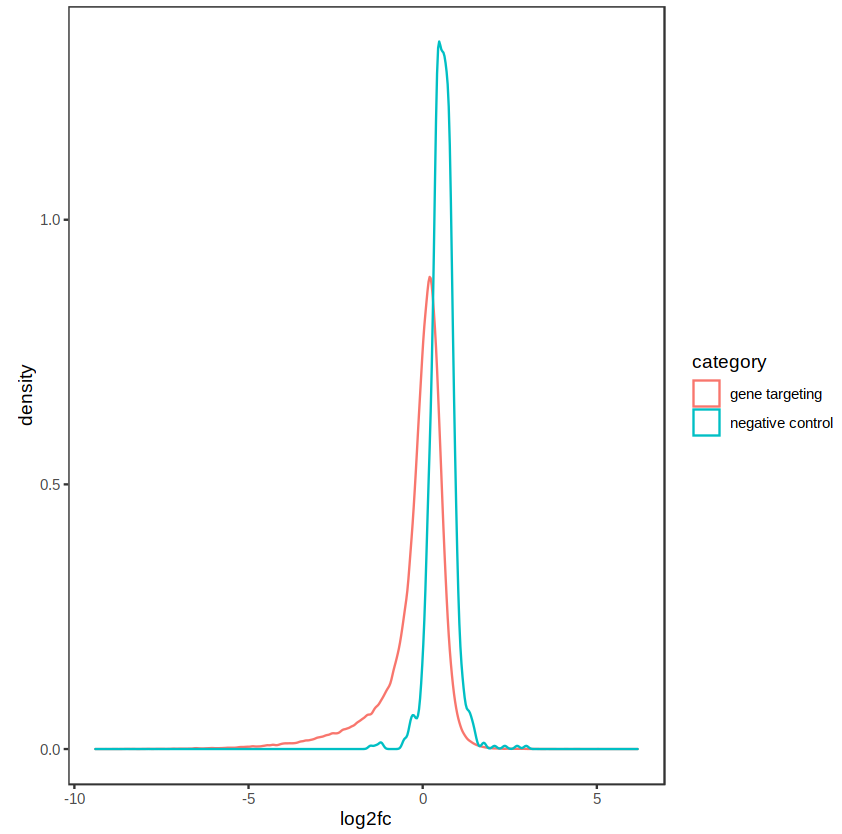
The distribution of fold-changes is similar regardless of design choice, which makes sense as there should be a high degree of overlap across cell lines:
design = ~ condition:
design = ~ cellType + condition:
Is one design strategy recommended over the other? Thanks in advance.
I am thinking about this section in the FAQ: http://www.bioconductor.org/packages/release/bioc/vignettes/DESeq2/inst/doc/DESeq2.html#multi-factor-designs
"Experiments with more than one factor influencing the counts can be analyzed using design formula that include the additional variables...By adding variables to the design, one can control for additional variation in the counts." I think controlling for cell line may be useful here.

Agree with Kevin, if you see samples clustering by celltype in PCA, that would be a good reason to use ~celltype + condition.