
Hello,
I am now using DiffBind (3.0.14) to analyze my ATAC-seq data. I have two groups of samples that have drastically different peaks numbers called by macs2. The cells before injury have around 16k peaks. The cells after injury have around 56k peaks. And the MA plot of non-normalized data generated by dba.plotMA(cls.dsq, method = DBA_DESEQ2, bNormalized = F, th = 0) shows that the injured samples have a much greater raw read density. Both the full library size normalization and background normalization methods did not alter this bias and the results are extremely unbalanced. The code I used for normalization is as below.
cls.cnt.normalized <- dba.normalize(cls.cnt, method = DBA_ALL_METHODS, normalize = DBA_NORM_LIB) #full library size normalization
#or
cls.cnt.normalized <- dba.normalize(cls.cnt, method = DBA_ALL_METHODS, normalize = DBA_NORM_NATIVE, background = TRUE) #background normalization
The analysis method I used is DBA_DESEQ2.
My first question is what is the best practice in DiffBind to deal with two data sets that have major changes in chromatin accessibility?
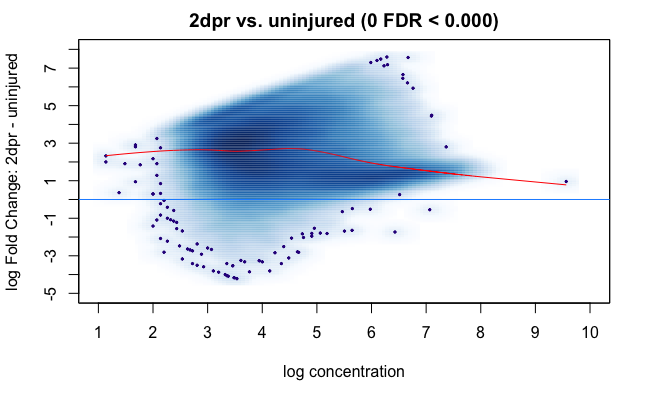
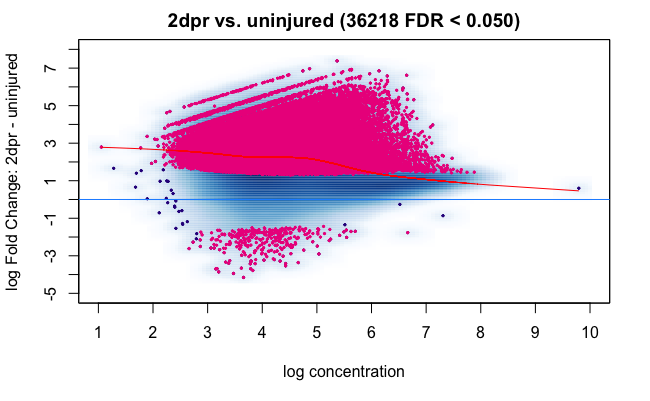
One way I have thought of to double-check whether the normalization is valid is to check the fragment counts (my data is paired-end) of promoter regions of highly expressed unchanged genes between uninjured and injured samples using their RNA-seq data. Now I have the promoter positions of the genes I want to look at but I am not sure how to retrieve the normalized count matrix of my samples in DiffBind. I only found RPKM of each sample at each consensus peak. And the RPKM value stays the same before and after dba.normalize call.
My second question is how is the RPKM calculated after the dba.count call? It is a value produced after library size and genomic interval length normalization of the raw fragment count, just like RNA-seq RPKM?
My final question is how can I retrieve the normalized count of all my samples with genomic locations (Chr Start End)? (I have tried
dsq.dds <- dba.analyze(cls.dsq, method = DBA_DESEQ2, bRetrieveAnalysis = T)
cnt.table <- counts(dsq.dds, normalized = T)
but the row names of the table are just numbers 1, 2, 3, 4, 5... lack of genomic location information.)
Many thanks in advance!
Best regards,
Shuwan

This could be relevant to you as well, normalization seems to be very offset here due to the fact that the 2dpr condition has a lot of regions with increased counts: Comment: How sensitive is DiffBind to Deseq2 assumptions ?
Maybe try to only use a subset of the regions for normalization.
See also Rory Stark's answer in this post : Normalization in DiffBind for Atac-Seq