
Hi,
I am attempting to examine alternative splicing with edgeR. I have moved through the analysis without obvious problems. However, when I investigate individual genes that show up as alternatively spliced (e.g. plotSpliceDGE) I notice that often what appears to be the same exon with the same start coordinate is repeated multiple times (see attached image for example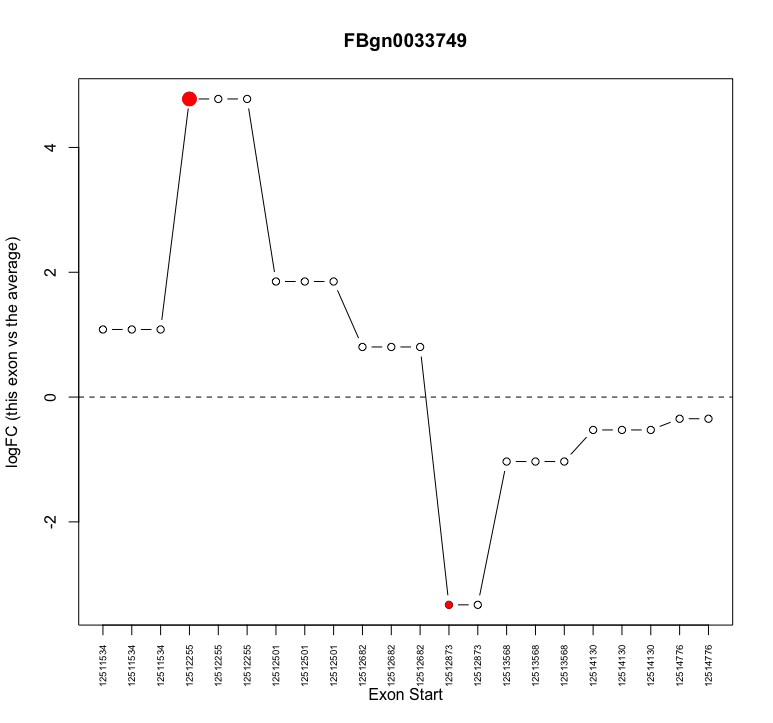
subjunc(index="mel_index",readfile1=reads1_2, readfile2=reads2_2, nthreads=8)
bamlist_splicing=list.files(pattern=".subjunc.sorted.BAM$")
counts_splicing <- featureCounts(bamlist_splicing, annot.ext="dmel-all-r6.38.gtf",
isGTFAnnotationFile=TRUE, GTF.featureType="exon",
useMetaFeatures=FALSE, allowMultiOverlap=TRUE, isPairedEnd=TRUE)
Counts_splicing_DGE <- DGEList(counts_splicing$counts, genes=counts_splicing$annotation, group=groups)
filter_keep_splicing <- filterByExpr(Counts_splicing_DGE)
Counts_splicing_DGE_filtered <- Counts_splicing_DGE[filter_keep_splicing,]
Counts_splicing_DGE_filtered_norm_factors <- calcNormFactors(Counts_splicing_DGE_filtered)
design <- model.matrix(~0+Group)
colnames(design) <- levels(Group)
Counts_splicing_DGE_filtered_norm_factors <- estimateDisp(Counts_splicing_DGE_filtered_norm_factors, design)
fit_splicing <- glmQLFit(Counts_splicing_DGE_filtered_norm_factors, design)
ControlvsMutant.F.1.splicing <- diffSpliceDGE(fit_splicing, geneid="GeneID", exonid="Start", contrast=contrasts[,"Y.F.1vsC.F.1"])
ControlvsMutant.F.1.splicing.results=topSpliceDGE(ControlvsMutant.F.1.splicing, test="Simes", n=Inf, FDR=1)
plotSpliceDGE(ControlvsMutant.F.1.splicing, geneid="FBgn0033749")
# include your problematic code here with any corresponding output
# please also include the results of running the following in an R session
sessionInfo( )
R version 4.0.4 (2021-02-15)
Platform: x86_64-apple-darwin17.0 (64-bit)
Running under: macOS Catalina 10.15.7
Matrix products: default
BLAS: /System/Library/Frameworks/Accelerate.framework/Versions/A/Frameworks/vecLib.framework/Versions/A/libBLAS.dylib
LAPACK: /Library/Frameworks/R.framework/Versions/4.0/Resources/lib/libRlapack.dylib
locale:
[1] en_US.UTF-8
attached base packages:
[1] stats4 parallel stats graphics grDevices utils datasets methods base
other attached packages:
[1] Rsubread_2.4.3 GenomicFeatures_1.42.2 GenomicRanges_1.42.0 GenomeInfoDb_1.26.4 ASpli_2.0.0
[6] AnnotationDbi_1.52.0 IRanges_2.24.1 S4Vectors_0.28.1 Biobase_2.50.0 BiocGenerics_0.36.0
[11] edgeR_3.32.1 limma_3.46.0
loaded via a namespace (and not attached):
[1] colorspace_2.0-0 ellipsis_0.3.1 biovizBase_1.38.0 htmlTable_2.1.0
[5] XVector_0.30.0 base64enc_0.1-3 dichromat_2.0-0 rstudioapi_0.13
[9] DT_0.17 bit64_4.0.5 fansi_0.4.2 xml2_1.3.2
[13] splines_4.0.4 cachem_1.0.4 knitr_1.31 Formula_1.2-4
[17] Rsamtools_2.6.0 cluster_2.1.1 dbplyr_2.1.0 png_0.1-7
[21] BiocManager_1.30.10 compiler_4.0.4 httr_1.4.2 backports_1.2.1
[25] lazyeval_0.2.2 assertthat_0.2.1 Matrix_1.3-2 fastmap_1.1.0
[29] htmltools_0.5.1.1 prettyunits_1.1.1 tools_4.0.4 igraph_1.2.6
[33] gtable_0.3.0 glue_1.4.2 GenomeInfoDbData_1.2.4 dplyr_1.0.5
[37] rappdirs_0.3.3 tinytex_0.30 Rcpp_1.0.6 vctrs_0.3.6
[41] Biostrings_2.58.0 rtracklayer_1.50.0 xfun_0.22 stringr_1.4.0
[45] lifecycle_1.0.0 ensembldb_2.14.0 statmod_1.4.35 XML_3.99-0.6
[49] zlibbioc_1.36.0 MASS_7.3-53.1 scales_1.1.1 BiocStyle_2.18.1
[53] BSgenome_1.58.0 VariantAnnotation_1.36.0 ProtGenerics_1.22.0 hms_1.0.0
[57] MatrixGenerics_1.2.1 SummarizedExperiment_1.20.0 AnnotationFilter_1.14.0 RColorBrewer_1.1-2
[61] yaml_2.2.1 curl_4.3 memoise_2.0.0 gridExtra_2.3
[65] ggplot2_3.3.3 UpSetR_1.4.0 biomaRt_2.46.3 rpart_4.1-15
[69] latticeExtra_0.6-29 stringi_1.5.3 RSQLite_2.2.4 checkmate_2.0.0
[73] BiocParallel_1.24.1 rlang_0.4.10 pkgconfig_2.0.3 matrixStats_0.58.0
[77] bitops_1.0-6 evaluate_0.14 lattice_0.20-41 purrr_0.3.4
[81] htmlwidgets_1.5.3 GenomicAlignments_1.26.0 bit_4.0.4 tidyselect_1.1.0
[85] plyr_1.8.6 magrittr_2.0.1 R6_2.5.0 generics_0.1.0
[89] Hmisc_4.5-0 DelayedArray_0.16.2 DBI_1.1.1 pillar_1.5.1
[93] foreign_0.8-81 survival_3.2-10 RCurl_1.98-1.3 nnet_7.3-15
[97] tibble_3.1.0 crayon_1.4.1 utf8_1.2.1 BiocFileCache_1.14.0
[101] rmarkdown_2.7 jpeg_0.1-8.1 progress_1.2.2 locfit_1.5-9.4
[105] grid_4.0.4 data.table_1.14.0 blob_1.2.1 digest_0.6.27
[109] tidyr_1.1.3 openssl_1.4.3 munsell_0.5.0 Gviz_1.34.1
[113] askpass_1.1

Hi Gordon,
Thanks so much!