
Hi,
I am confused about the annotation of downstream region and it's priority in ChIPseeker. By default downstream defined from TTS to +3kb, and in my understanding, peaks overlapped with this region will be annotated as downstream if the priority of downstream is set to the first. However, nothing changed when I do that. There is my example:
Firstly, I annotated data using default priority(c("Promoter", "5UTR", "3UTR", "Exon", "Intron", "Downstream", "Intergenic"))
peakAnnolist <- lapply(samplefiles, annotatePeak, TxDb = txdb, tssRegion = c(-1000, 1000), annoDb = "org.Mm.eg.db")
# part results
GRanges object with 2 ranges and 12 metadata columns:
seqnames ranges strand | annotation geneChr geneStart geneEnd geneLength geneStrand geneId
<Rle> <IRanges> <Rle> | <character> <integer> <integer> <integer> <integer> <integer> <character>
[1] chr11 28692672-28694373 * | Exon (ENSMUST00000146554.1/72818, exon 3 of 3) 11 28681564 28693276 11713 1 72818
[2] chr11 28697582-28698686 * | Distal Intergenic 11 28681564 28693276 11713 1 72818
transcriptId distanceToTSS ENSEMBL SYMBOL GENENAME
<character> <numeric> <character> <character> <character>
[1] ENSMUST00000146554.1 11108 ENSMUSG00000084966 2810471M01Rik RIKEN cDNA 2810471M01 gene
[2] ENSMUST00000146554.1 16018 ENSMUSG00000084966 2810471M01Rik RIKEN cDNA 2810471M01 gene
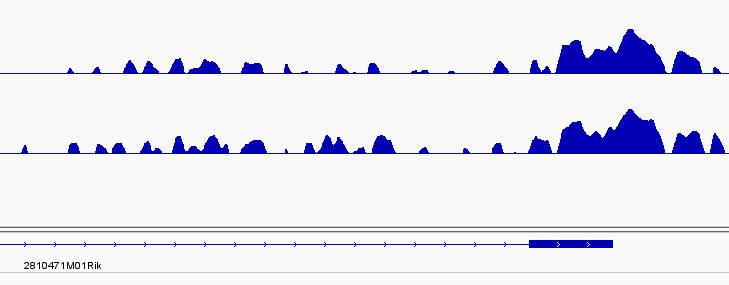
Peak(28692672-28694373) was overlapped with the geneEnd site and annotated as Exon(consistent with the IGV visualization). But I hoped that peaks like this could be annotated as downstream. Then I set the genomicAnnotationPriority to c("Promoter","Downstream", "5UTR", "3UTR", "Exon", "Intron", "Intergenic"):
peakAnnolist <- lapply(samplefiles, annotatePeak, TxDb = txdb, tssRegion = c(-1000, 1000), annoDb = "org.Mm.eg.db",
genomicAnnotationPriority = c("Promoter","Downstream", "5UTR", "3UTR", "Intron", "Intergenic"))
# part results
GRanges object with 2 ranges and 12 metadata columns:
seqnames ranges strand | annotation geneChr geneStart geneEnd geneLength geneStrand geneId
<Rle> <IRanges> <Rle> | <character> <integer> <integer> <integer> <integer> <integer> <character>
[1] chr11 28692672-28694373 * | Exon (ENSMUST00000146554.1/72818, exon 3 of 3) 11 28681564 28693276 11713 1 72818
[2] chr11 28697582-28698686 * | Distal Intergenic 11 28681564 28693276 11713 1 72818
transcriptId distanceToTSS ENSEMBL SYMBOL GENENAME
<character> <numeric> <character> <character> <character>
[1] ENSMUST00000146554.1 11108 ENSMUSG00000084966 2810471M01Rik RIKEN cDNA 2810471M01 gene
[2] ENSMUST00000146554.1 16018 ENSMUSG00000084966 2810471M01Rik RIKEN cDNA 2810471M01 gene
You can see that the result was same as the previous. And not only this peak, all the annotation results did not change. I also tried to remove the Exon from genomicAnnotationPriority, however, I failed again as it was just become 'Distal Intergenic'.
What should I do to solve this problem?
Thanks!