
Hi bioconductor team,
I'm analyzing atac-seq data with 4 genotypes and two biological replicate for each of them (8 samples overall). I used macs2 callpeak for peak calling and then used the .bam files and .narrowPeak files to apply Diffbind and get the binding matrix. Also, I'm working with DiffBind_2.14.0. I need to mention that the bam files have gone through the quality control and reads with MAPQ<30 are removed.
I know that all my .narrowPeak files contain peaks in HOXa region (I annotated every single of them and hoxa regions overlap within all samples). However, I cannot find this region in the total binding matrix. I even set minOverlap=1 to get total number of unique peaks after merging overlapping ones, and then annotated all the regions but HOXa was not found.
So, I believe that DiffBind is filtering some of the regions somehow. My question is that what filtering steps the DiffBind is applying in order to generate the binding matrix?
dbaObj <- dba(sampleSheet=cellSamples,peakCaller="narrow",
config=data.frame(doBlacklist=FALSE, doGreylist=FALSE, singleEnd=FALSE, yieldSize=5000000,
bUsePval=TRUE,th=0.01), scoreCol=5, filter=0)
When I annotated all the regions from dba$merged I could find Skap2 but not HOXa. Below is a screenshot from uploading bw files to igv, showing the peaks for Skap2 and hoxa.
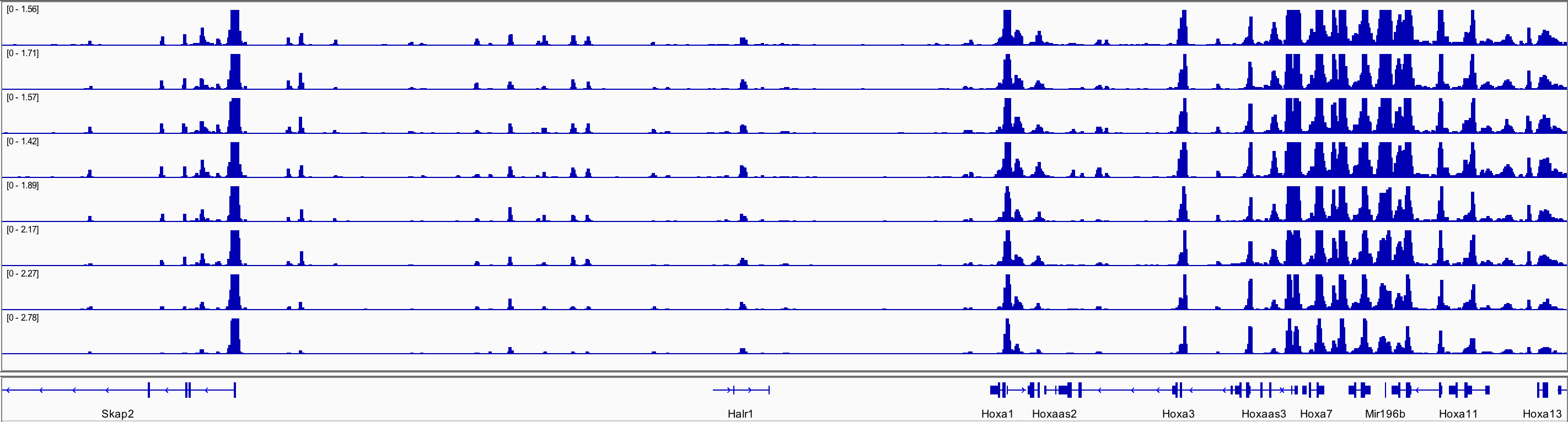
Hoxa genes go from chr6:52,105,000-52,217,000, and this interval is available in all .narrowPeak files, but how come DiffBind remove it when making the binding matrix? If the dba() function does any filtering can you please explain it to me?
Many thanks,
Shiva.

Thank you for your response. Yes I found the regions related to "hoxa" when I used
dba.peakset(). So why do the regions fromdbaObj$bindinganddba.peakset()differ? (the dim of both data frames are the same though)Ultimately I need the binding matrix to do DB analysis later. Based on my understanding,
dba.count()usesdbaObj$bindingmatrix in order to count reads in binding site intervals, and thedbaObj$bindingmiss some regions. What is the reason for that? And which one is correct?Thanks again.
Everything that is supposed to be there is there! The internal
$bindingmatrix uses a type of factor representation of chromosome names, so the chromosome numbered 6 is not "chr6".dba.peakset()withbRetrieve=TRUEtakes care of converting chromosome name strings (with the help of the internal vector$chrmap).Thank you for helping me figuring this out! The matrix from dba.peakset() seems to be the correct one. Thanks!