Entering edit mode

I need to map mouse Ensembl gene ID's to their corresponding Entrez Gene ID's. In the process of reviewing cases of multi-mapping ID's, I came across a few examples where the wrong Entrez ID is assigned to a corresponding Ensembl ID.
For Example:
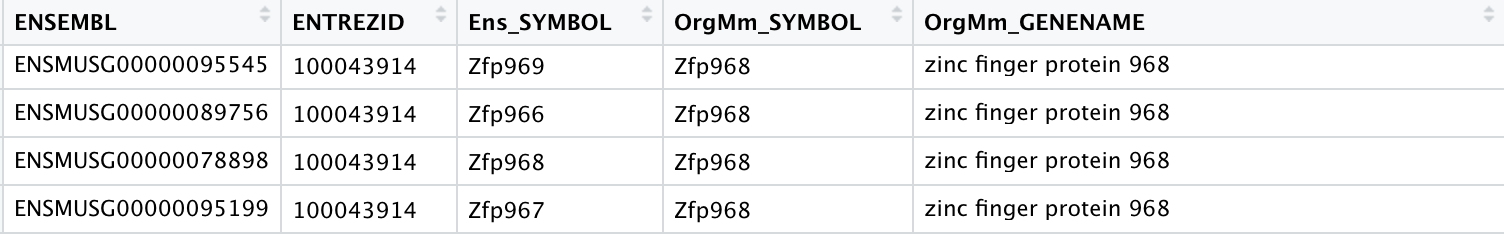
Are all recognized by NCBI, and in most cases the NCBI gene page recognizes the correct Ensembl annotation:
- Zfp966 Is Entrez ID 667962, which NCBI recognizes as Ensembl ID: ENSMUSG00000089756
- Zfp968 Is Entrez ID 100043914, which NCBI recognizes as Ensembl ID: ENSMUSG00000078898
- Zfp967 Is Entrez ID 100303732, which NCBI recognizes as Ensembl ID: ENSMUSG00000095199
Another examples include:
- Ccl27a, Entrez ID 20301 which NCBI recognizes as Ensembl ID: ENSMUSG00000073888
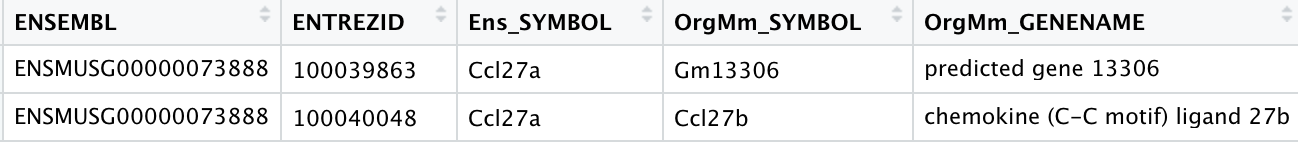
and
- Ndufb4, Entrez ID 68194 which NCBI recognizes as Ensembl ID: ENSMUSG00000022820

If there is an error in my query (below), please let me know.
## Get Unique Ensembl Gene ID's from differential expression analysis
ens_mm_gid <- deg_master %>%
filter(grepl("ENSMUS", gene_id))%>%
pull("gene_id") %>% unique()
## Query org.Mm.eg.db with Ensembl Gene ID Keys
ens_mm_entrez <- AnnotationDbi::select(
org.Mm.eg.db,
columns = c("ENTREZID","SYMBOL","GENENAME"),
keys = ens_mm_gid,
keytype = "ENSEMBL"
)
## Evaluate cases where 2 or more Ensembl ID's are assigned the same Entrezid
ens_mm_entrez %>%
inner_join(
mouse_an %>% select(gene_id, Ens_SYMBOL=SYMBOL),
by=c(ENSEMBL="gene_id")
)%>%
filter(!is.na(ENTREZID)) %>%
group_by(ENTREZID) %>%
filter(n() > 1) %>%
arrange(ENTREZID)%>%
select(
ENSEMBL, ENTREZID, Ens_SYMBOL,
OrgMm_SYMBOL=SYMBOL, OrgMm_GENENAME=GENENAME
) %>%
View()
sessionInfo( )
R version 4.0.5 (2021-03-31)
Platform: x86_64-apple-darwin17.0 (64-bit)
Running under: macOS Catalina 10.15.7
Matrix products: default
BLAS: /System/Library/Frameworks/Accelerate.framework/Versions/A/Frameworks/vecLib.framework/Versions/A/libBLAS.dylib
LAPACK: /Library/Frameworks/R.framework/Versions/4.0/Resources/lib/libRlapack.dylib
locale:
[1] en_US.UTF-8/en_US.UTF-8/en_US.UTF-8/C/en_US.UTF-8/en_US.UTF-8
attached base packages:
[1] stats4 grid parallel stats graphics grDevices utils datasets methods base
other attached packages:
[1] openxlsx_4.2.4 dplyr_1.0.7 org.Mm.eg.db_3.12.0 AnnotationDbi_1.52.0 IRanges_2.24.1 S4Vectors_0.28.1 Biobase_2.50.0 ROntoTools_2.18.0
[9] Rgraphviz_2.34.0 KEGGgraph_1.50.0 KEGGREST_1.30.1 boot_1.3-28 graph_1.68.0 BiocGenerics_0.36.1 synapser_0.10.89 edgeR_3.32.1
[17] limma_3.46.0
loaded via a namespace (and not attached):
[1] Rcpp_1.0.7 locfit_1.5-9.4 lattice_0.20-44 tidyr_1.1.3 png_0.1-7 Biostrings_2.58.0 assertthat_0.2.1 packrat_0.6.0
[9] utf8_1.2.1 R6_2.5.0 RSQLite_2.2.7 httr_1.4.2 pillar_1.6.1 zlibbioc_1.36.0 rlang_0.4.11 rstudioapi_0.13
[17] blob_1.2.1 RCurl_1.98-1.3 bit_4.0.4 compiler_4.0.5 pkgconfig_2.0.3 pack_0.1-1 tidyselect_1.1.1 tibble_3.1.2
[25] codetools_0.2-18 XML_3.99-0.6 fansi_0.5.0 crayon_1.4.1 bitops_1.0-7 lifecycle_1.0.0 DBI_1.1.1 magrittr_2.0.1
[33] zip_2.2.0 cli_3.0.1 stringi_1.7.3 cachem_1.0.5 PythonEmbedInR_0.7.80 XVector_0.30.0 ellipsis_0.3.2 generics_0.1.0
[41] vctrs_0.3.8 tools_4.0.5 bit64_4.0.5 glue_1.4.2 purrr_0.3.4 fastmap_1.1.0 memoise_2.0.0

I should note that when I query org.Mm.eg.db using the gene Symbols associated with my differential expression data I find a 1:1 correspondence between a symbol and an Entrez ID, at least for the set of genes that matter in our study. I was always taught that database accession's were more reliable unique identifiers than gene symbols and should be preferred in bioinformatic analyses. Perhaps this is a position I aught to reconsider?