Entering edit mode


I'm applying clustal-omega multiple sequence alignment through msa package for aligning 600 dna covid sequences and it is running from yesterday till now so almost 24 hours and didn't finish yet. Is it normal ? it consumes most of RAMs 8Gb and 31% cpu. However, I read in papers that clustal-omega can align >50000 seq in few hours.
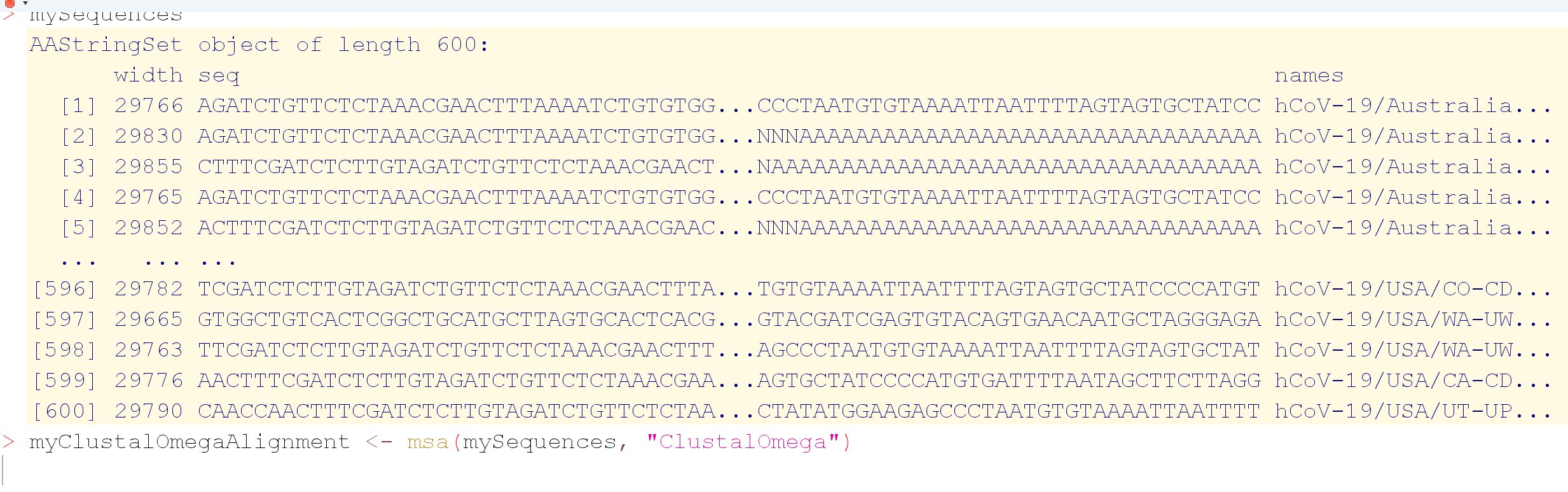
From your screendump it seems that your DNA sequences are containend in an AAStringSet instead of a DNAStringSet, which makes some difference:
# Aligning 100 DNA sequences of length 2000
class(seq.dna)
# [1] "DNAStringSet"
# attr(,"package")
# [1] "Biostrings"
system.time(seq.dna.msa <- msa(seq.dna, method="ClustalOmega"))
# using Gonnet
# user system elapsed
# 41.70 6.42 48.16
# Aligning the same 100 DNA sequences contained in an AAStringSet
class(seq.aa)
# [1] "AAStringSet"
# attr(,"package")
# [1] "Biostrings"
system.time(seq.aa.msa <- msa(seq.aa, method="ClustalOmega"))
# using Gonnet
# user system elapsed
# 55.50 6.13 61.69
So you may want to read your sequences with readDNAStringSet().
How can I apply this and what are the resources required to get benefit from multi-threading?
msaClustalW() has a parameter threads= to speed up the alignment, but specifying it seems to make no difference, at least on Windows.
system.time(seq.dna.msa <- msa(seq.dna, method="ClustalOmega", threads=4))
# using Gonnet
# user system elapsed
# 42.03 6.34 48.49
Use of this site constitutes acceptance of our User Agreement and Privacy Policy.