Entering edit mode

I'm working with DESeq2 to make a DE analysis between samples in two different conditions. During the analysis, I identified a batch effect due to the sequencing time modelled as a covariate in the design formula. From the differential expression (Wald test), I was able to retrieve a good number of significant genes (~100) but the LogFC range looks not reliable going from -30 to +30.
- What could be the cause of these extreme large values and how can I solve the problem? I tried to use lfcShrink() to re-estimate the logFC but I'm not sure that is sufficient to achieve reliable results.
- My second question is about the design of the model. Is it reasonable to add covariates to the model also if they don't show a strong effect on the data (looking PCA or clustering)?
Follow the code used for the analysis. Thanks for your help!
dds <- DESeqDataSetFromMatrix(count, coldata, design = ~ Group + Condition)
keep <- rowSums(counts(dds)) >= 10
dds <- dds[keep,]
dds <- DESeq(dds)
res <- results(dds, contrast=c("Condition","dis","hea"))
res <- res[which(res$padj <0.05),]

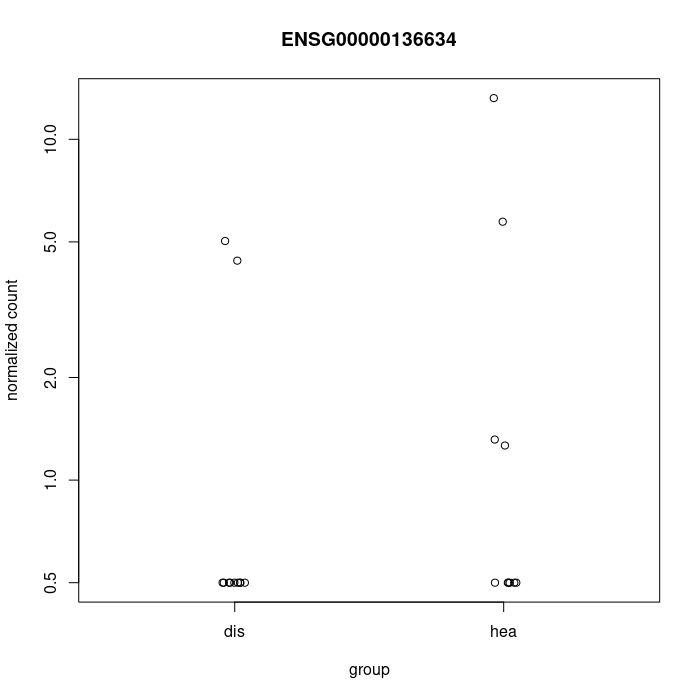
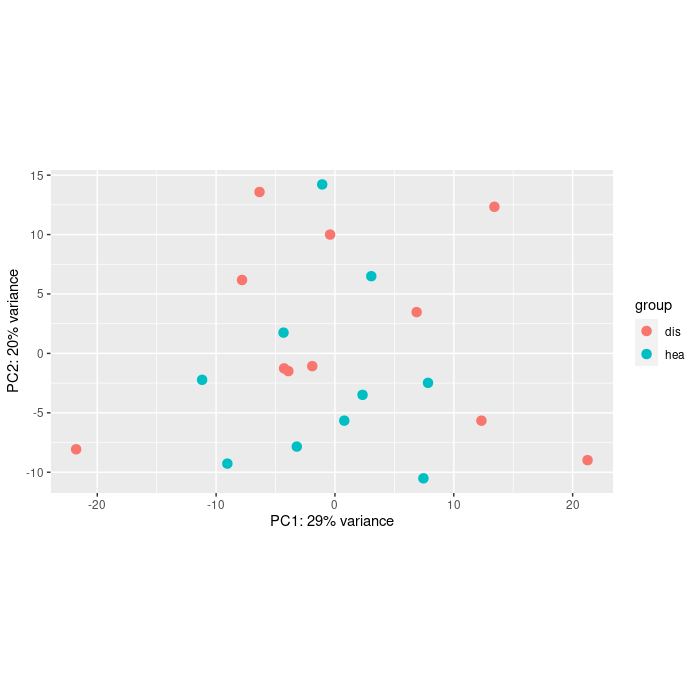
You could at least show some counts of the genes with large FCs. Also the PCA would help.