
Hello,
Recently, I am learning the RNA-seq analysis, especially for PCA in order to check if the control and treatment can separate based on the treatment condition. I checked online for preparing the data before performing the PCA. I got an idea that, LCPM calculated from raw count matrix can be used as the PCA input and I did it the my first PCA method below. The resulting PCA can separate between control and treatment (see the figure here: 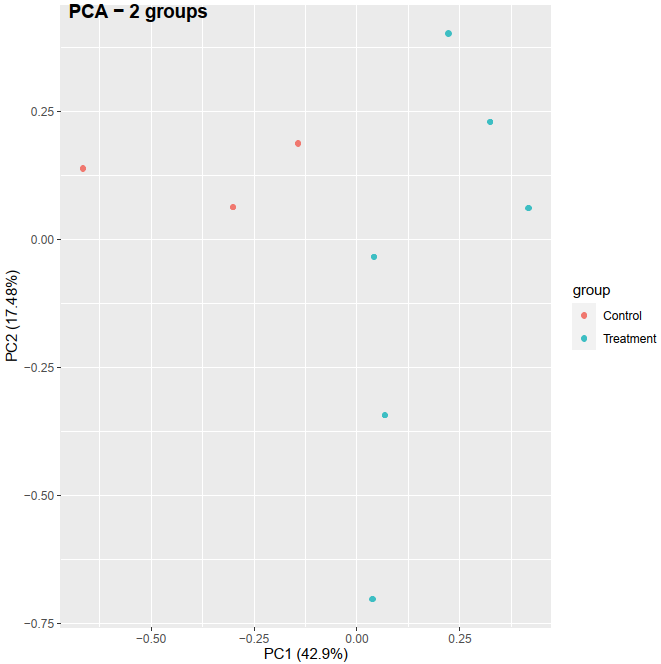
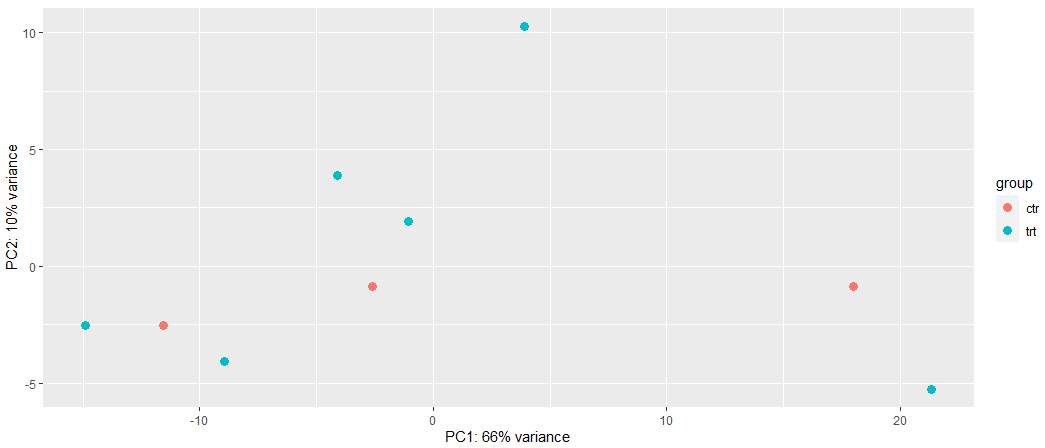
Can you give suggestions which method (LCPM or VST) should I used in this case? Why are different in PCA based on the different data transformation? In this case which data transformation method should I use? Or based on the PCA methods I tried, can it indicate that my RNA-seq data is hard to separate based on treatment from control?
Any suggestion are appreciated!
Some key codes are listed below.
# Method 1: PCA based on LCPM
y <- readDGE(files, columns = c(1, 3))
lcpm <- cpm(y, log = TRUE)
dat <- t(lcpm)
pca_res <- prcomp(dat, scale. = TRUE)
autoplot(pca_res, data = dat_org, colour = 'group')
# Method 2: VST
dds <- DESeqDataSetFromMatrix(countData = cts,
colData = coldat,
design = ~ condition)
vsd <- vst(dds, blind = FALSE)
plotPCA(vsd, intgroup = c("condition"))

This is not like-for-like. In the first PCA you use all genes and even scale them while the second one uses the top 500 most variable genes wihout scaling. These differences are stronger than the vst/logcpm difference. Vst or logcpm, in my experience it really does not matter too much but be sure to do the PCA correctly as in here it's inconsistent.
ATpoint Thanks so much. Now, I understand that 'scale' and 'top 500 most variables' play an import role in the different result in my case.
May I ask one more question, should I do the scaling operation before performing PCA in the method 01? I noted that, by doing scaling, control and treatment can separate in PC1, while without doing scaling, then control and treatment can separate in PC2.
As you mentioned "be sure to do the PCA correctly", can you help give the suggestions what is the corrected way to do PCA for RNA-Seq data?
Thanks.
Please follow the edgeR user guide on PCA/MDS exactly unless you have expert knowledge to confidently change anything they suggested. DESeq2 is another option, excellent user guide as well.
Thanks ATpoint I will do that.