Entering edit mode

Hello, I have been using txImport to add up all the transcripts to get one TPM value per gene. When I add up genes manually in excel, it doesn't always match tximport's TPM value. Why is tximport TPM's sometimes less than the expected value I calculated in Excel? Here is a comparison of the TPMs:
Gene- [Excel] vs [TxImport Result]
Aanat - 0 vs 0
Aatk - 0.29 vs 0.17
Abca1 - 3.28 vs 3.28
Abca4 - 0.25 vs. 0.22
Abca2 - 3.2 vs 2.75
Abcb7 - 0.95 vs 0.95
Manual Count in Excel:
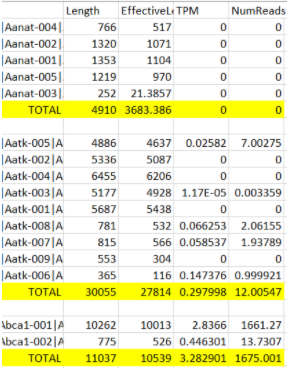
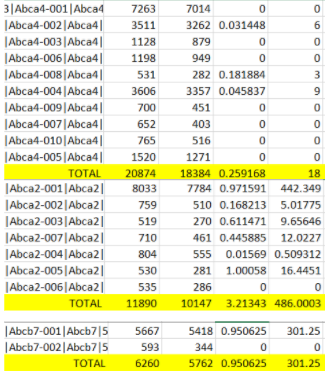
TxImport Result:
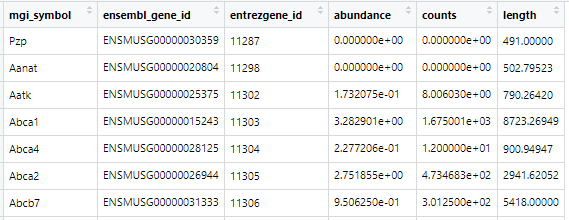
Here is my code for reference
#load in data
#make file path to file in C:\Users\cathe\Desktop
files <- file.path("C:", "Users", "cathe", "Desktop", "GSM3106294_MEF_1_quant2.sf.txt", fsep="/")
#get gene ID's and TX names
library(TxDb.Mmusculus.UCSC.mm10.knownGene)
txdb <- TxDb.Mmusculus.UCSC.mm10.knownGene
txdb
k <- keys(txdb, keytype = "GENEID")
length(k)
df <- select(txdb, keys = k, keytype = "GENEID", columns = "TXNAME")
tx2gene <- df[, 2:1] # tx ID, then gene ID
head(tx2gene)
#run tximport
txi.salmon <- tximport(files, type = "salmon", tx2gene = tx2gene)
txi.salmon
#view tximport output that has gene level quantification
head(txi.salmon$counts)
#convert the tximport object to a data frame for downstream operations
txi.salmon <- as.data.frame(txi.salmon)
#move rownames with the gene ID to a column using data.table package
setDT(txi.salmon, keep.rownames = "entrezgene_id")
#convert entrezid from character to int data type downstream operations
txi.salmon$entrezgene_id <- as.integer(txi.salmon$entrezgene_id)
#get gene symbol for entrez id in another data frame
library(biomaRt)
mart <- useMart('ensembl', dataset = 'mmusculus_gene_ensembl')
entrezid2symbol <-
getBM(
attributes = c(
'ensembl_gene_id',
'entrezgene_id', 'mgi_symbol'),
filters = 'entrezgene_id',
values = txi.salmon$entrezgene_id,
mart = mart)
entrezid2symbol <- as.data.frame(entrezid2symbol)
#merge both tximport salmon output and entrezid2symbol to include gene symbols
salmonids <- merge(txi.salmon,entrezid2symbol,by=c("entrezgene_id"))
write.table(salmonids, file="output.txt", sep = "\t", row.names=FALSE, col.names=FALSE, quote = FALSE)
sessionInfo( )
> sessionInfo()
R version 4.0.5 (2021-03-31)
Platform: x86_64-w64-mingw32/x64 (64-bit)
Running under: Windows 10 x64 (build 18363)
Matrix products: default
locale:
[1] LC_COLLATE=English_United States.1252 LC_CTYPE=English_United States.1252
[3] LC_MONETARY=English_United States.1252 LC_NUMERIC=C
[5] LC_TIME=English_United States.1252
attached base packages:
[1] parallel stats4 stats graphics grDevices utils datasets methods base
other attached packages:
[1] data.table_1.14.0 TxDb.Mmusculus.UCSC.mm10.knownGene_3.10.0
[3] GenomicFeatures_1.42.3 GenomicRanges_1.42.0
[5] GenomeInfoDb_1.26.7 biomaRt_2.46.3
[7] org.Mm.eg.db_3.12.0 AnnotationDbi_1.52.0
[9] IRanges_2.24.1 S4Vectors_0.28.1
[11] Biobase_2.50.0 BiocGenerics_0.36.0
[13] readr_1.4.0 tximport_1.18.0
[15] MOFA2_1.3.0
loaded via a namespace (and not attached):
[1] bitops_1.0-6 matrixStats_0.58.0 bit64_4.0.5
[4] filelock_1.0.2 RColorBrewer_1.1-2 progress_1.2.2
[7] httr_1.4.2 tools_4.0.5 utf8_1.2.1
[10] R6_2.5.0 HDF5Array_1.18.1 uwot_0.1.10
[13] DBI_1.1.1 colorspace_2.0-0 rhdf5filters_1.2.0
[16] withr_2.4.2 tidyselect_1.1.0 prettyunits_1.1.1
[19] bit_4.0.4 curl_4.3 compiler_4.0.5
[22] basilisk.utils_1.2.2 xml2_1.3.2 DelayedArray_0.16.3
[25] rtracklayer_1.49.5 scales_1.1.1 askpass_1.1
[28] rappdirs_0.3.3 Rsamtools_2.6.0 stringr_1.4.0
[31] basilisk_1.2.1 XVector_0.30.0 pkgconfig_2.0.3
[34] MatrixGenerics_1.2.1 dbplyr_2.1.1 fastmap_1.1.0
[37] rlang_0.4.10 rstudioapi_0.13 RSQLite_2.2.5
[40] generics_0.1.0 jsonlite_1.7.2 BiocParallel_1.24.1
[43] dplyr_1.0.5 RCurl_1.98-1.3 magrittr_2.0.1
[46] GenomeInfoDbData_1.2.4 Matrix_1.3-2 Rcpp_1.0.6
[49] munsell_0.5.0 Rhdf5lib_1.12.1 fansi_0.4.2
[52] reticulate_1.18 lifecycle_1.0.0 stringi_1.5.3
[55] SummarizedExperiment_1.20.0 zlibbioc_1.36.0 rhdf5_2.34.0
[58] Rtsne_0.15 plyr_1.8.6 BiocFileCache_1.14.0
[61] grid_4.0.5 blob_1.2.1 ggrepel_0.9.1
[64] forcats_0.5.1 crayon_1.4.1 lattice_0.20-41
[67] Biostrings_2.58.0 cowplot_1.1.1 hms_1.0.0
[70] pillar_1.6.0 reshape2_1.4.4 XML_3.99-0.6
[73] glue_1.4.2 vctrs_0.3.7 gtable_0.3.0
[76] openssl_1.4.3 purrr_0.3.4 tidyr_1.1.3
[79] assertthat_0.2.1 cachem_1.0.4 ggplot2_3.3.3
[82] tibble_3.1.0 pheatmap_1.0.12 GenomicAlignments_1.26.0
[85] memoise_2.0.0 corrplot_0.84 ellipsis_0.3.1
