
This might be a bit long, please bare with me. I'm conducting a differential expression analysis using limma - voom. My comparison is regarding response vs non-response to a cancer drug. However, I'm not getting any DE genes, absolute zeros. Someone here once recommended not to use contrast matrix for such a simple task, so I did that but then I get almost all 18 thousand genes as DE, so that's not good either. I don't know what is the problem here. The counts matrix and metadata matrix have the same samples with the exact same order. I filtered my counts matrix from noise genes earlier. I did everything according to this guide: https://bioconductor.org/packages/release/workflows/vignettes/RNAseq123/inst/doc/limmaWorkflow.html
<h5>Note: This problem is not only in this dataset, this is just one of many. So the problem is probably in my code.</h5>Let me show you the code and the results I get:
Code:
d0 = DGEList(counts)
group = metadata$Response
d0$samples$group = group
geneid = rownames(d0)
d0$genes = geneid
d0 = calcNormFactors(d0, method = 'TMM')
dim(d0)
## Design
design = model.matrix(~ 0 + Response, metadata) %>% as.data.frame()
colnames(design)[c(1,2)] = c('NoResponse','Response')
## Contrast
contrast = makeContrasts(NoResp_VS_Resp = NoResponse - Response, levels = colnames(design))
Voom <- voom(d0, design, plot = TRUE)
vfit <- lmFit(Voom, design = design)
vfit <- contrasts.fit(vfit , contrasts = contrast)
efit <- eBayes(vfit)
plotSA(efit, main = 'final model: Mean-Variance trend')
summary(decideTests(efit))
deg <- topTable(efit,
coef = 'NoResp_VS_Resp',
p.value = 0.05,
adjust.method = 'fdr',
number = Inf)
Now you might ask how does the Voom and the eBayes plots look like, here they are:
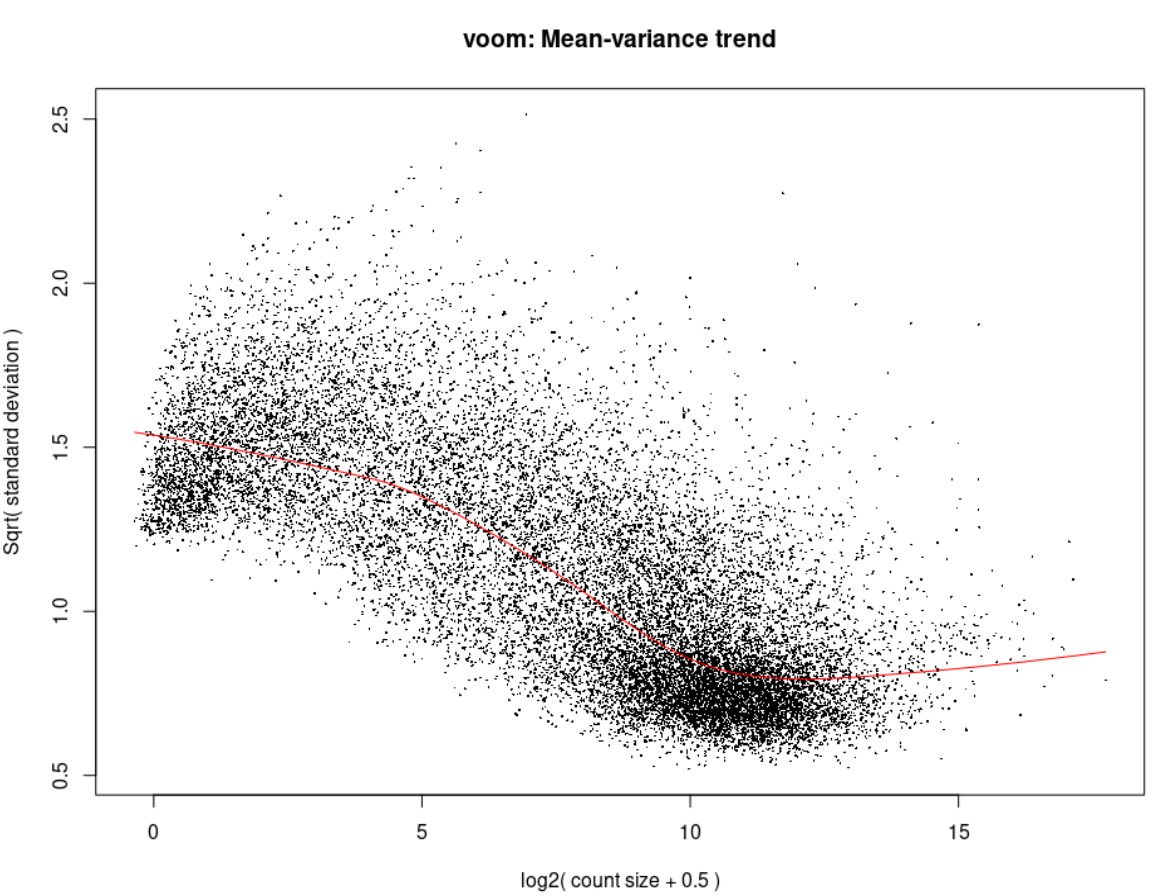
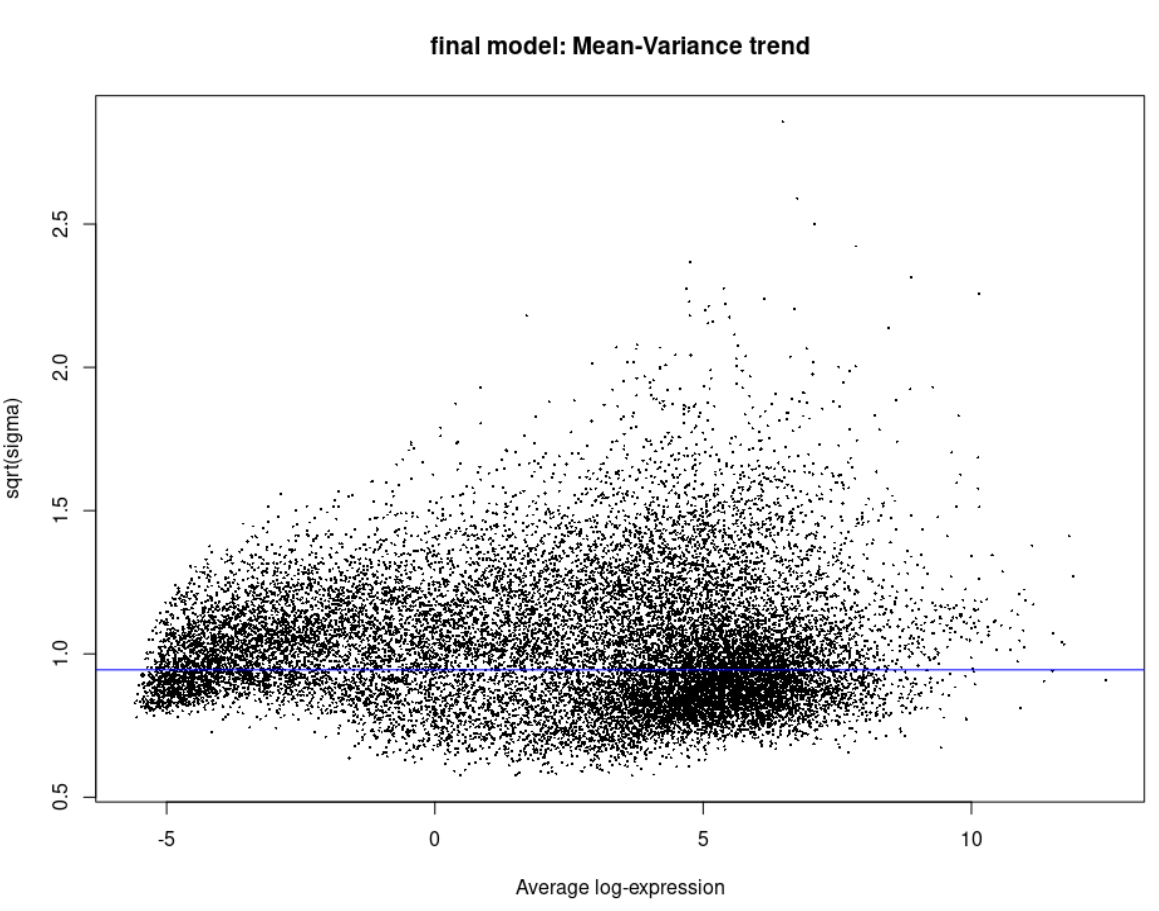
And here is the summary(decideTests(efit)) results when using contrast matrix:

And this is without contrast matrix: ( Almost 17 thousand significant genes, out of 18 thousand)

Can anyone help me figure out what is the problem ? If any additional info is needed I'll add it!

I see.. one final question, regarding gene filtering.
Could you tell me why this isn't working?
Gives this error :
Error in object[[a]][i, , drop = FALSE] : incorrect number of dimensionsBTW, with human data like yours, I would almost always use
voomWithQualityWeightsinstead ofvoomin order to allow for poor quality samples or outliers. That will generally increase power to detect DE genes.Thanks a ton, your help is realy appreciated!
That would be because you set
d0$genesto be a vector instead of a data.frame, so that thegenescomponent can't be subsetted by rows. In other words, yourd0isn't a valid DGEList object. Seehelp("DGEList-class").To fix this, just delete the lines
They're unnecessary and they make
d0unsubsettable.Later on, you convert
designinto a data.frame. That won't cause any errors, but it is supposed to remain a matrix.