
I have a cds object file for which I want to do single cell RNA seq analysis (make UMAP and identify cell types) on using monocle and garnett software. I was trying to check what marker genes are in my cds file.
cds1 <- readRDS('example_cds.RDS') head(colData(cds1)) #shows annotations on each column as: cell (character), size factor (numeric), n.umi (numeric), perc_mitochondrial_umis (numeric), scrublet_score (numeric), scrublet_call (character), num_genes_expressed (integer) head(rowData(cds1)) #shows annotations on each row as: gene_short_name (character), id (character), chromosome (character), bp1 (integer), bp2 (integer), gene_strand (character), num_cells_expressed (integer)
library(org.Mm.eg.db)
marker_file_path <- "C:/Users/[my username here]/Downloads/kidney_marker_genes.txt"
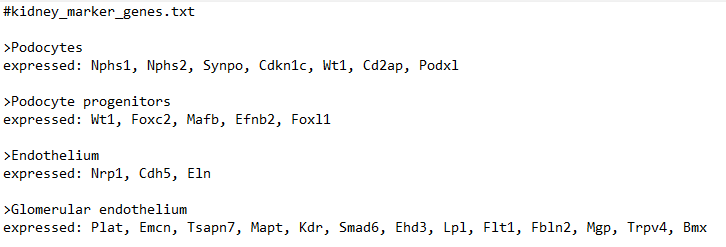
marker_check <- check_markers(cds1, marker_file_path, db=org.Mm.eg.db, cds_gene_id_type = "SYMBOL", marker_file_gene_id_type = "SYMBOL")
plot_markers(marker_check)
However, I'm getting an error (see attached screenshot)
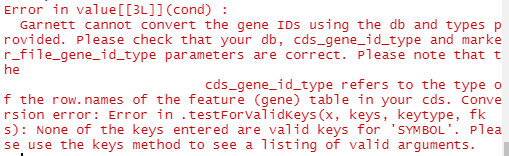
Do you have any suggestions on how I can troubleshoot this step?
