
Hi,
I am interested in fitting gene expression in two different stages of development (RNA-seq counts, quantile-normalised, and log1p-transformed). Based on visualising a scatter plot of the two stages, I have noticed that variance is not as high for higher levels of gene expression, compared for genes with lower values of gene expression. I have learned this is called heteroscedasticity and I would like to account for it in order to model properly.
hco <- hcor_ab[[1]] # this is a data frame with two columns 'a' and 'b' corresponding to two stages, and 3641 rows corresponding to genes
jsd_value <- 0 # a value that I need in the plot below
hco_lm <- lm(b ~ a, data = hco)
library(ggpointdensity)
ggplot(data = hco,mapping = aes(x=a,y=b)) +
labs(title = names(hcor_ab)[1] ) +
geom_pointdensity()+
stat_smooth(method = "lm", se = FALSE, color = "red") +
theme_minimal()+
scale_color_viridis()+
annotate(
"text", x = Inf, y = -Inf, hjust = 1, vjust = 0,
label = paste(
"Equation: y =", sprintf("%.2f", coef(summary(lm(b ~ a, data = hco)))[2, 1]), "x +",
sprintf("%.2f", coef(summary(lm(b ~ a, data = hco)))[1, 1]), "\n",
"Rel. J-S Divergence = ", jsd_value
)
)
See below the resulting plot of this code.
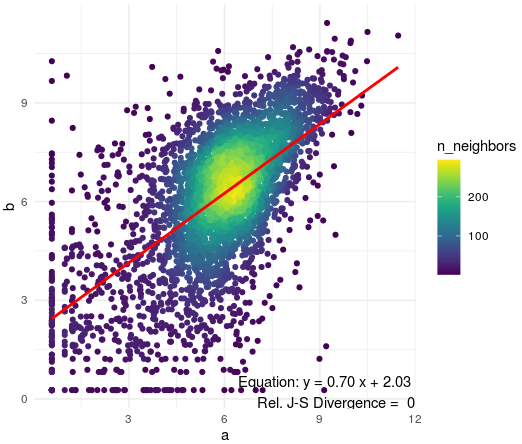
To account for heteroscedasticity, I have been advised to use the inverse of the expected sampling variance as weights, and to compute bootstraps for this. But I am at a loss on how to proceed.
One extra thing to consider: I have to implement this inside a larger function that takes the two-condition data frame as input, to be run by other end-users, so in principle it would be in my best interest to rely just on the data that is fed to the function (the hco object) and no other a-priori information.
Thank you very much in advance!

Hi Michael,
Sorry for the late reply. Thank you for pointing this out to me. While it is true that this would satisfy my example case, I will not be able to control the source of the input data in future cases by other users; therefore I was thinking if there is another way to account for heteroscedasticity just with the input data (i.e. counts prior to log1p-transformation and quantile normalisation, but not necessarily with pre-computed bootstraps available).
Thank you for your help and your time,
Alberto
Maybe if you look at data where you have the sampling variability you can model it in datasets where you don't. Minimally you should expect Poisson variability, where variance = mean.