
Dear Community,
based on a ongoing analysis of a microarray dataset, with affymetrix platform HTA 2.0, i would like to ask some very quick and specific questions regarding the first steps of preprocessing/normalization/annotation, as this platform is relatively "more complex" that other arrays. In detail,
Based on a similar post that i have created recently: C: Preprocessing of Human Gene 2.0 ST microarrays with oligo R package and annotati
1) Regarding the rma normalization with the oligo R package, in the option :
data.norm <- rma(affy.cels, target="core")
Again the option "core", is the more appropriate way to move forward, as i would like to perform downstream DE analysis, and this option contains essentially more "safely annotated genes" with the utilization of trancript clusters?
2) Regarding the annotation process, after normalization, still the following functions would be ok, and except the initial R package pd.hta.2.0 and utilization of the R package affycoretools ??:
# firstly remove the control probes with getMainProbes() # and for a quick annotation process: library(hta20transcriptcluster.db) eset.rma <- annotateEset(data.norm, hta20transcriptcluster.db)
3) Finally, the function paCalls from oligo package, is also plaucible for the HTA.2.0 platform ?
Thank you in advance,
Efstathios

Thank you James for your confirmation !!!
Just your brief opinion/comment that is more relevant for my 3rd question regarding paCalls and the possibility of non-specific intensity filtering: because this affymetrix platform contains negative control probes, do you believe that a non-specific intensity filtering, based on negative controls and as described in the page 111 with agilent case study
(https://www.bioconductor.org/packages/devel/bioc/vignettes/limma/inst/doc/usersguide.pdf) ?
or again this specific approach is innapropriate for my specific platform ?
Or if my notion is correct, in order to access the negative control probes in my case study, i have to change my pre-processing steps from above ?
You can't use paCalls regardless.
So there isn't a method for HTAFeatureSets, and so paCalls Just Won't Work (TM).
You could hypothetically come up with your own version, but it's not clear how you would extend that to the transcript level, and trying to analyze an HTA array at the probeset level is, in my opinion, way more trouble than it is worth.
Anyway, there is the easy way to do things (that is probably OK most of the time), and the hard way to do things (that might arguably be a better thing to do, all things equal). The easy thing to do would be to do a plot and eyeballometrically decide what cutoff to use if you want to exclude probesets that you think are not expressed.
> z <- getMainProbes("pd.hta.2.0") > plot(density(exprs(eset)[as.character(z[z[,2] %in% 1,1]),1]), main = "Probeset distribution", ylim = c(0,0.6)) > for(i in c(2,7)) lines(density(exprs(eset)[as.character(z[z[,2] %in% i,1]),1]), lty = if(i == 2) 2 else 3) > legend("topright", c("Main","Antigenomic","Intronic"), lty = 1:3, bty="n")The plot of my data, using the intronic probesets, shows that a cutoff of somewhere around 2.5 would separate the arguably expressed from unexpressed probesets, at least for the first sample. You could make this plot for all the samples to get a better idea.
Also note how the antigenomic probes have a really wide distribution - this shows that, given a high enough GC content, an Affy probe will bind to pretty much anything (the high GC probes are pretty much GCGCGCGCGCGC, and Affy says these things don't exist in nature, so they are binding to things that they are not complementary to).
An alternative method would be to bin your data based on GC content of the probes, and then exclude those that were below the set of antigenomic probesets with corresponding GC content. Which would be boring and tedious to do, and doing the simple thing is probably OK anyway.
Thank you very much for your explanation on why paCalls does not work for HTAFeatureSets and for describing an alternative method on how to find an appropriate cutoff to separate expressed from unexpressed probesets.
I am working with HTA 2.0 as well and tried to follow your advice of simply plotting the distribution for main, antigenomic and intronic probesets. However, I was only able to plot the distribution for the main probesets since according to the platform design info pd.hta.2.0, they all seem to be of type "1" (main) or "NA":
> eset <- rma(rawData) > z <- getMainProbes("pd.hta.2.0") > table(z$type)I thought that according to problems filtering antigenomic probes from HTA 2.0 , there also should be some of type 2-7. Is that correct?
I am using affycoretools version 1.48.0 and pd.hta.2.0 version 3.12.1.
I would be very grateful for any help!
Hi Prof. James, I'm confused with the procedures used for filtering the unexpressed genes mentioned here. Why is the cutoff based on intronic probesets? And what is the purpose for the antigenomic probesets here for filtering? Here is my results. Thanks in advance!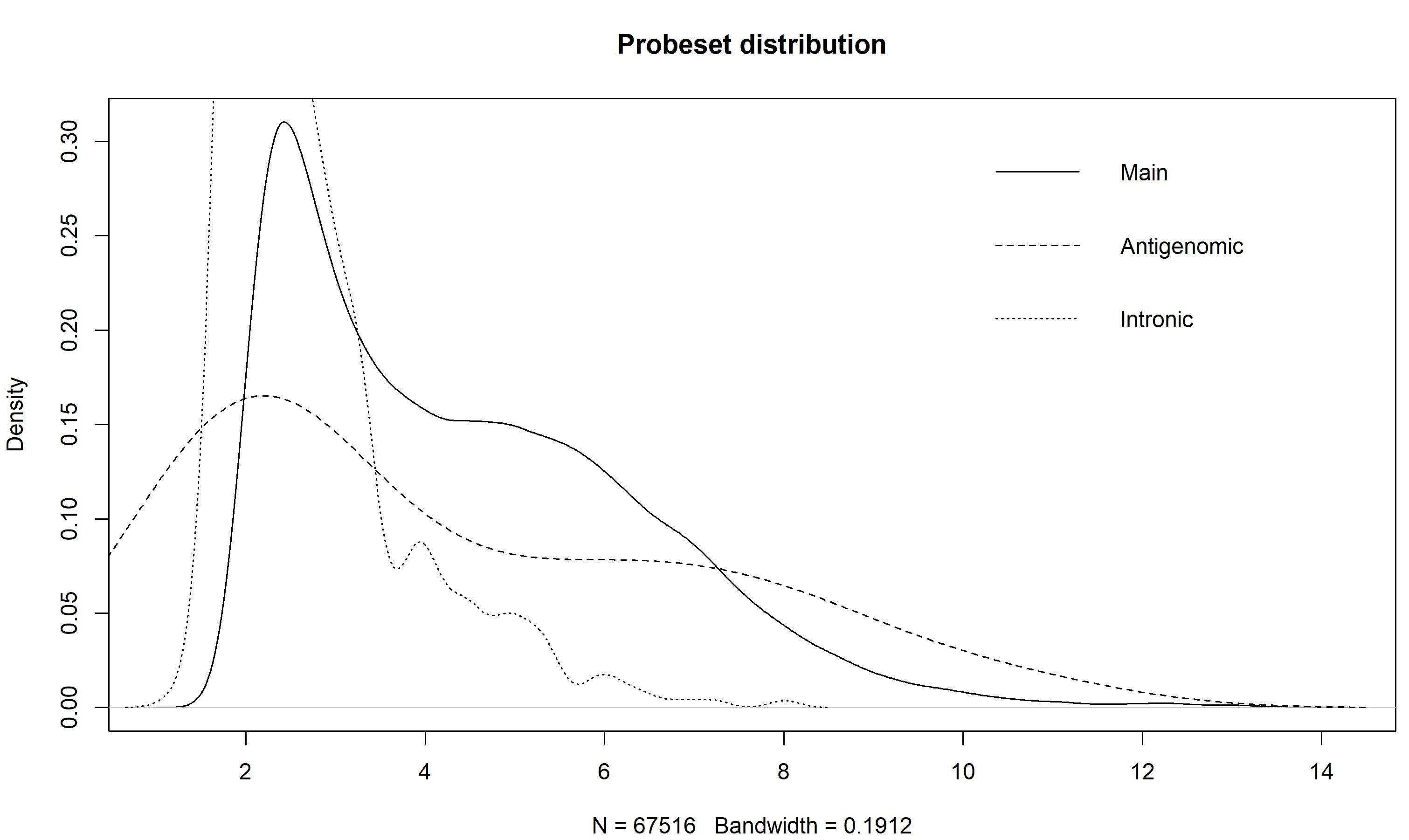
Please don't add comments onto years-old posts. Instead make a new post.
okay sir, the new post is here (Methods for removing unexpressed probes of Human Transcriptome Array HTA 2.0). Looking forward to your reply.