
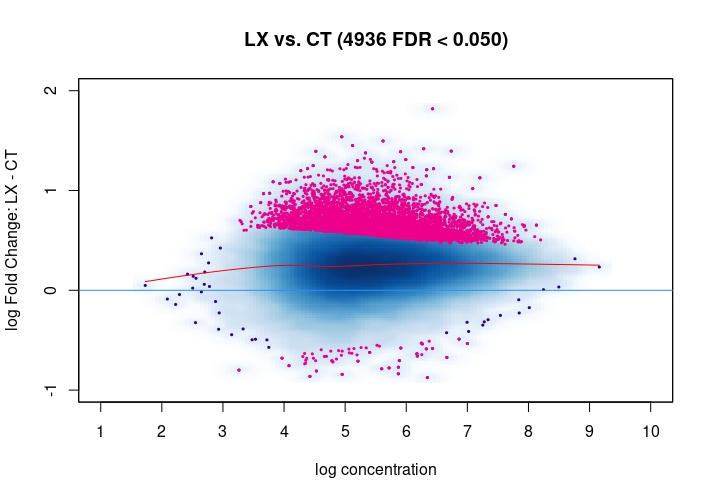
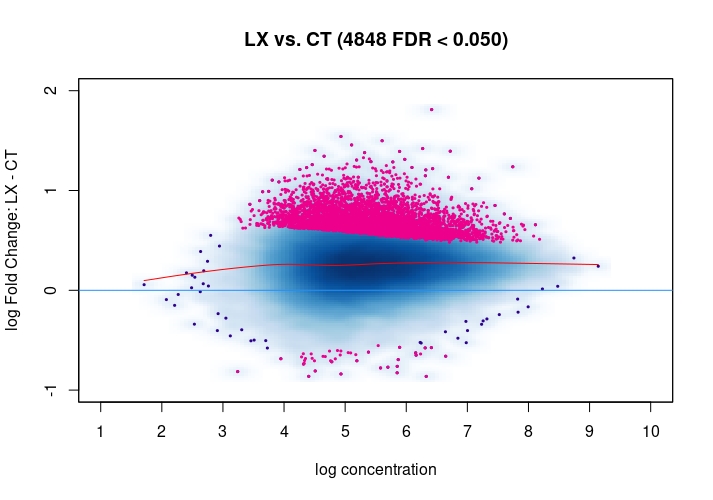
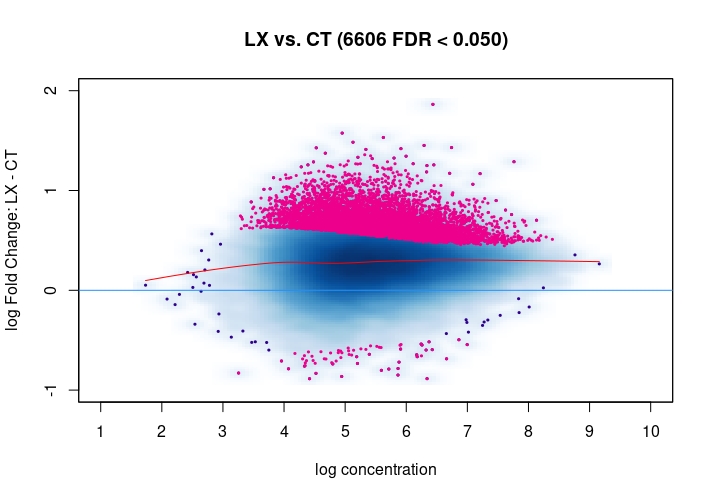
Hi, I have been using DiffBind_3.0.7 for determining Differential accessible sites. I have CT and KD samples, 3 replicates each, and we expect a large scale opening of the chromatin in the KD. After calling Peaks with MACS2 and running the DiffBind steps, I used 3 different Normalization commands to see how or whether they differ significantly in terms of numbers of differentially open sites . I read the Normalization part of your Vignette, and it mentions that one of the recommendations for atac-seq is actually the second option I use. I was wondering if the first option would be a valid way. Also, a full library size normalization ( option 3 ) should be similar to option 2, but I get much more sites. Would you recommend I stick with Option 2 ? I am attaching the MA plots ( Options 1,2 and 3 )
Also, if in a situation where I do no expect widespread changes of chromatin accessibility, would you recommend RLE on Reads in Peaks normalization?
Thank you, Debayan
Code should be placed in three backticks as shown below
mod_atacseq_data.norm1 <- dba.normalize(mod_atacseq_data, method=DBA_DESEQ2, normalize=DBA_NORM_LIB, library=DBA_LIBSIZE_BACKGROUND, background=TRUE)
mod_atacseq_data.norm2 <- dba.normalize(mod_atacseq_data, method=DBA_DESEQ2, normalize=DBA_NORM_NATIVE, library=DBA_LIBSIZE_BACKGROUND, background=TRUE)
mod_atacseq_data.norm3 <- dba.normalize(mod_atacseq_data, method=DBA_DESEQ2, normalize=DBA_NORM_LIB, library=DBA_LIBSIZE_FULL, background=FALSE)
#Contrasts#
mod_atacseq_data.norm1.ctst <- dba.contrast(mod_atacseq_data.norm1, minMembers=3, categories=DBA_CONDITION)
mod_atacseq_data.norm2.ctst <- dba.contrast(mod_atacseq_data.norm2, minMembers=3, categories=DBA_CONDITION)
mod_atacseq_data.norm3.ctst <- dba.contrast(mod_atacseq_data.norm3, minMembers=3, categories=DBA_CONDITION)
#Differential Analysis#
mod_atacseq_data.results1 <- dba.analyze(mod_atacseq_data.norm1.ctst, method=DBA_DESEQ2)
mod_atacseq_data.results2 <- dba.analyze(mod_atacseq_data.norm2.ctst, method=DBA_DESEQ2)
mod_atacseq_data.results3 <- dba.analyze(mod_atacseq_data.norm3.ctst, method=DBA_DESEQ2)
mod_atacseq_data.DB1 <- dba.report(mod_atacseq_data.results1, method=DBA_DESEQ2, bCalled=T, bCounts=T)
mod_atacseq_data.DB2 <- dba.report(mod_atacseq_data.results2, method=DBA_DESEQ2, bCalled=T, bCounts=T)
mod_atacseq_data.DB3 <- dba.report(mod_atacseq_data.results3, method=DBA_DESEQ2, bCalled=T, bCounts=T)
bindingdata1=as.data.frame(mod_atacseq_data.DB1)
bindingdata2=as.data.frame(mod_atacseq_data.DB2)
bindingdata3=as.data.frame(mod_atacseq_data.DB3)
sum(bindingdata1$Fold>0)
4882
> sum(bindingdata1$Fold<0)
54
> sum(bindingdata2$Fold>0)
4806
> sum(bindingdata2$Fold<0)
42
> sum(bindingdata3$Fold>0)
6543
> sum(bindingdata3$Fold<0)
63
dba.plotMA(mod_atacseq_data.results1)
dba.plotMA(mod_atacseq_data.results2)
dba.plotMA(mod_atacseq_data.results3)
sessionInfo( )
R version 4.0.2 (2020-06-22)
Platform: x86_64-pc-linux-gnu (64-bit)
Running under: CentOS Linux 7 (Core)
Matrix products: default
BLAS/LAPACK: /usr/lib64/libopenblasp-r0.3.3.so
locale:
[1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C LC_TIME=en_US.UTF-8 LC_COLLATE=en_US.UTF-8
[5] LC_MONETARY=en_US.UTF-8 LC_MESSAGES=en_US.UTF-8 LC_PAPER=en_US.UTF-8 LC_NAME=C
[9] LC_ADDRESS=C LC_TELEPHONE=C LC_MEASUREMENT=en_US.UTF-8 LC_IDENTIFICATION=C
attached base packages:
[1] stats4 parallel stats graphics grDevices utils datasets methods base
other attached packages:
[1] csaw_1.24.3 DiffBind_3.0.7 SummarizedExperiment_1.20.0
[4] MatrixGenerics_1.2.0 matrixStats_0.57.0 Hmisc_4.4-2
[7] Formula_1.2-4 survival_3.1-12 lattice_0.20-41
[10] impute_1.64.0 EnsDb.Hsapiens.v79_2.99.0 ensembldb_2.14.0
[13] AnnotationFilter_1.14.0 GenomicFeatures_1.42.1 AnnotationDbi_1.52.0
[16] Biobase_2.50.0 GenomicRanges_1.42.0 GenomeInfoDb_1.26.1
[19] IRanges_2.24.0 S4Vectors_0.28.0 BiocGenerics_0.36.0
[22] forcats_0.5.0 purrr_0.3.4 readr_1.4.0
[25] tidyr_1.1.2 tidyverse_1.3.0 data.table_1.13.2
[28] stringr_1.4.0 dplyr_1.0.2 ggfortify_0.4.11
[31] ggplot2_3.3.2 tibble_3.0.4 RColorBrewer_1.1-2
[34] pheatmap_1.0.12 readxl_1.3.1
loaded via a namespace (and not attached):
[1] tidyselect_1.1.0 RSQLite_2.2.1 htmlwidgets_1.5.2 grid_4.0.2
[5] BiocParallel_1.24.1 munsell_0.5.0 base64url_1.4 systemPipeR_1.24.2
[9] withr_2.3.0 colorspace_2.0-0 Category_2.56.0 knitr_1.30
[13] rstudioapi_0.13 bbmle_1.0.23.1 GenomeInfoDbData_1.2.4 mixsqp_0.3-43
[17] hwriter_1.3.2 bit64_4.0.5 coda_0.19-4 batchtools_0.9.14
[21] vctrs_0.3.5 generics_0.1.0 xfun_0.19 BiocFileCache_1.14.0
[25] R6_2.5.0 apeglm_1.12.0 invgamma_1.1 locfit_1.5-9.4
[29] rsvg_2.1 bitops_1.0-6 DelayedArray_0.16.0 assertthat_0.2.1
[33] scales_1.1.1 nnet_7.3-14 gtable_0.3.0 rlang_0.4.9
[37] genefilter_1.72.0 splines_4.0.2 rtracklayer_1.50.0 lazyeval_0.2.2
[41] broom_0.7.2 brew_1.0-6 checkmate_2.0.0 BiocManager_1.30.10
[45] yaml_2.2.1 modelr_0.1.8 backports_1.2.0 RBGL_1.66.0
[49] tools_4.0.2 ellipsis_0.3.1 gplots_3.1.1 Rcpp_1.0.5
[53] plyr_1.8.6 base64enc_0.1-3 progress_1.2.2 zlibbioc_1.36.0
[57] RCurl_1.98-1.2 prettyunits_1.1.1 rpart_4.1-15 openssl_1.4.3
[61] ashr_2.2-47 haven_2.3.1 ggrepel_0.8.2 cluster_2.1.0
[65] fs_1.5.0 magrittr_2.0.1 reprex_0.3.0 truncnorm_1.0-8
[69] mvtnorm_1.1-1 SQUAREM_2020.5 amap_0.8-18 ProtGenerics_1.22.0
[73] hms_0.5.3 xtable_1.8-4 XML_3.99-0.5 emdbook_1.3.12
[77] jpeg_0.1-8.1 gridExtra_2.3 compiler_4.0.2 biomaRt_2.46.0
[81] bdsmatrix_1.3-4 KernSmooth_2.23-17 V8_3.4.0 crayon_1.3.4
[85] htmltools_0.5.0 GOstats_2.56.0 geneplotter_1.68.0 lubridate_1.7.9.2
[89] DBI_1.1.0 dbplyr_2.0.0 MASS_7.3-51.6 rappdirs_0.3.1
[93] ShortRead_1.48.0 Matrix_1.2-18 cli_2.2.0 pkgconfig_2.0.3
[97] GenomicAlignments_1.26.0 numDeriv_2016.8-1.1 foreign_0.8-80 xml2_1.3.2
[101] annotate_1.68.0 XVector_0.30.0 AnnotationForge_1.32.0 rvest_0.3.6
[105] VariantAnnotation_1.36.0 digest_0.6.27 graph_1.68.0 Biostrings_2.58.0
[109] cellranger_1.1.0 htmlTable_2.1.0 edgeR_3.32.0 GSEABase_1.52.0
[113] GreyListChIP_1.22.0 curl_4.3 Rsamtools_2.6.0 gtools_3.8.2
[117] rjson_0.2.20 lifecycle_0.2.0 jsonlite_1.7.1 askpass_1.1
[121] limma_3.46.0 BSgenome_1.58.0 fansi_0.4.1 pillar_1.4.7
[125] httr_1.4.2 GO.db_3.12.1 glue_1.4.2 png_0.1-7
[129] bit_4.0.4 Rgraphviz_2.34.0 stringi_1.5.3 blob_1.2.1
[133] DESeq2_1.30.0 latticeExtra_0.6-29 caTools_1.18.0 memoise_1.1.0
[137] DOT_0.1 irlba_2.3.3

Please show the MA-plots, you can use the image button to upload images (the one right of the
Codebutton).Sorry, I have just uploaded them. Thanks!
Just my two cents: Basically these plots look all the same. I would even argue that they are all offset towards the LX condition which is probably more of a normalization artifact than true biology. The data look kind of symmetric like in any other ATAC-seq experiment I've seen, supported by the local regression trend, and the fact that the bulk of the very high-count regions (far right) is also offset towards LX makes it even more questionable whether this is really true biology in terms of a general shift. I would strongly recommend to explore alternative normalization strategies and also check for confounders, e.g. FRiPs so whether data quality is notable different between data. See e.g. the
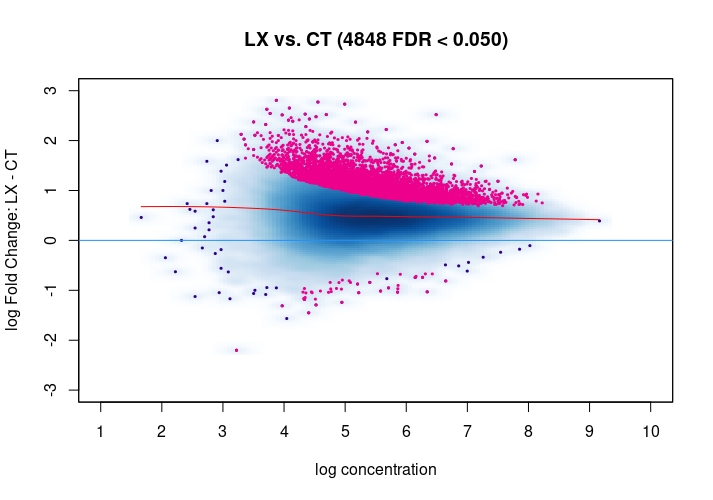
csawvignette for suggestions. I am not a DiffBind user so not sure what exactly the above strategies do, therefore suggestingcsawto explore data (edit:) as an independent approach.Also, it would be good to see the non-normalized MA plot!
Thanks a lot for your suggestion, I will try csaw as an independent approach. I have attached the non-normalized MA plot. There does appear to be deviation towards the y=0 line upon normalization, but as you mention there is a definite offset towards LX in the normalized MA plot