
Hello,
I am a new user of DESeq2 and I am having trouble with log2FoldChange calculation with latest version of DESeq2 package. DEGs reported in pairwise comparison using Wald Tests show completely abnormally high values of log2FoldChange (up to -30 and +20) I managed to recalculate an approximate of what would be a decent log2FoldChange per gene using
plotCounts
function and re-calculating log2foldChange for each gene as log2(mean(counts_condB))-log2(mean(counts_condA)). I know it is not exactly the way it is done in DESeq2 but I expect something close in term of order of magnitude. Values calculated this way where way lower and seemed to make more sense (at least there is no abs(log2FoldChange) above 10).
I can't really find what I am doing wrong here. I saw on a previous post that someone had a similar issue that was solved removing the parallel=T argument od DESeq function which I tried without sucess. You'll find below my code and infos.
dds<-DESeq2::DESeqDataSetFromMatrix(countData=sample_cts,colData=subset,design=~Mortalite+Temperature+Mortalite:Temperature+Batch+Manip)
keep<-rowSums(counts(dds) >= 10) >= 3
dds<-dds[keep,]
dds$Mortalite <- relevel(dds$Mortalite, ref = "ok")
dds_Wald<-DESeq(dds,parallel=T,BPPARAM=MulticoreParam(30),test="Wald")
fullresnames<-resultsNames(dds_Wald)
res<-DESeq2::results(dds_Wald,name=fullresnames[2],alpha=0.01,lfcThreshold = 2)
head(data.frame(res) %>% subset(padj<0.01) %>% arrange(desc(abs(log2FoldChange))))
baseMean log2FoldChange lfcSE stat pvalue padj
TRINITY_DN10323_c0_g1_i1 11.97128 -30 4.869130 -5.750514 8.897229e-09 3.794748e-06
TRINITY_DN1044_c0_g1_i7 14.07173 -30 4.865994 -5.754220 8.704285e-09 3.794748e-06
TRINITY_DN2267_c2_g1_i12 18.34856 -30 4.863310 -5.757396 8.542143e-09 3.794748e-06
TRINITY_DN2356_c0_g1_i7 44.30817 -30 4.863334 -5.757367 8.543604e-09 3.794748e-06
TRINITY_DN2590_c0_g1_i5 29.76859 -30 4.863410 -5.757277 8.548156e-09 3.794748e-06
TRINITY_DN2614_c0_g1_i5 30.91978 -30 3.814330 -7.340740 2.124169e-13 2.687073e-10
plotCounts(dds_Wald, "TRINITY_DN10323_c0_g1_i1", intgroup="Mortalite")
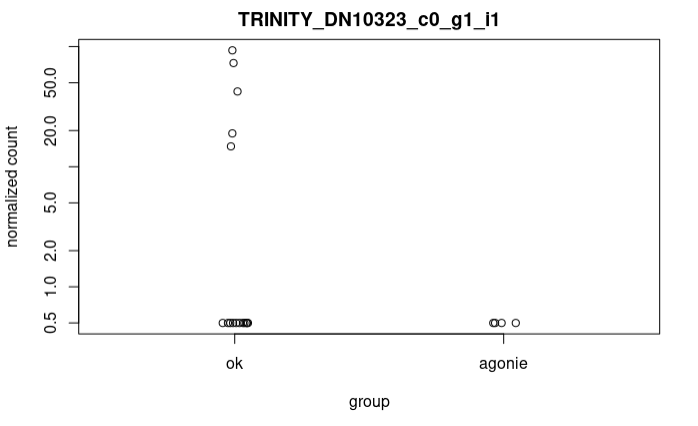
As you can see the log2FoldChange value is strange regarding the counts. If I calculate the log2FoldChange as described before :
x<-plotCounts(dds_Wald, "TRINITY_DN10323_c0_g1_i1" , intgroup="Mortalite",returnData = T) %>% group_by(Mortalite) %>% summarize(mean=mean(count))
log2(x$mean[x$Mortalite=="agonie"])-log2(x$mean[x$Mortalite=="ok"])
[1] -4.950851
sessionInfo()
R version 4.0.3 (2020-10-10)
Platform: x86_64-pc-linux-gnu (64-bit)
Running under: Ubuntu 20.04.2 LTS
Matrix products: default
BLAS: /usr/lib/x86_64-linux-gnu/blas/libblas.so.3.9.0
LAPACK: /usr/lib/x86_64-linux-gnu/lapack/liblapack.so.3.9.0
Random number generation:
RNG: L'Ecuyer-CMRG
Normal: Inversion
Sample: Rejection
locale:
[1] LC_CTYPE=fr_FR.UTF-8 LC_NUMERIC=C LC_TIME=fr_FR.UTF-8 LC_COLLATE=fr_FR.UTF-8 LC_MONETARY=fr_FR.UTF-8
[6] LC_MESSAGES=fr_FR.UTF-8 LC_PAPER=fr_FR.UTF-8 LC_NAME=C LC_ADDRESS=C LC_TELEPHONE=C
[11] LC_MEASUREMENT=fr_FR.UTF-8 LC_IDENTIFICATION=C
attached base packages:
[1] stats4 parallel stats graphics grDevices utils datasets methods base
other attached packages:
[1] forcats_0.5.0 stringr_1.4.0 purrr_0.3.4 readr_1.4.0
[5] tibble_3.0.4 tidyverse_1.3.0 gplots_3.1.1 GO.db_3.11.4
[9] AnnotationDbi_1.50.3 rgsepd_1.20.2 goseq_1.40.0 geneLenDataBase_1.24.0
[13] BiasedUrn_1.07 apeglm_1.10.0 pheatmap_1.0.12 vsn_3.56.0
[17] BiocParallel_1.22.0 randomForest_4.6-14 MLSeq_2.6.0 caret_6.0-86
[21] ggplot2_3.3.2 lattice_0.20-41 tidyr_1.1.2 dplyr_1.0.2
[25] tseries_0.10-47 DESeq2_1.28.1 SummarizedExperiment_1.18.2 DelayedArray_0.14.1
[29] matrixStats_0.57.0 Biobase_2.48.0 GenomicRanges_1.40.0 GenomeInfoDb_1.24.2
[33] phyloseq_1.32.0 edgeR_3.30.3 limma_3.44.3 Biostrings_2.56.0
[37] XVector_0.28.0 IRanges_2.22.2 S4Vectors_0.26.1 BiocGenerics_0.34.0
loaded via a namespace (and not attached):
[1] sSeq_1.26.0 tidyselect_1.1.0 RSQLite_2.2.1 grid_4.0.3 pROC_1.16.2
[6] munsell_0.5.0 codetools_0.2-18 preprocessCore_1.50.0 withr_2.3.0 colorspace_2.0-0
[11] GOSemSim_2.14.2 NLP_0.2-1 knitr_1.30 rstudioapi_0.13 TTR_0.24.2
[16] trinotateR_1.0 rrvgo_1.0.2 slam_0.1-47 bbmle_1.0.23.1 GenomeInfoDbData_1.2.3
[21] bit64_4.0.5 rhdf5_2.32.4 coda_0.19-4 vctrs_0.3.5 generics_0.1.0
[26] ipred_0.9-9 xfun_0.19 BiocFileCache_1.12.1 R6_2.5.0 rsvd_1.0.3
[31] locfit_1.5-9.4 bitops_1.0-6 assertthat_0.2.1 promises_1.1.1 scales_1.1.1
[36] nnet_7.3-15 gtable_0.3.0 affy_1.66.0 timeDate_3043.102 rlang_0.4.8
[41] genefilter_1.70.0 splines_4.0.3 rtracklayer_1.48.0 ModelMetrics_1.2.2.2 wordcloud_2.6
[46] broom_0.7.2 modelr_0.1.8 BiocManager_1.30.10 yaml_2.2.1 reshape2_1.4.4
[51] backports_1.1.10 GenomicFeatures_1.40.1 httpuv_1.5.4 quantmod_0.4.17 tools_4.0.3
[56] lava_1.6.8.1 gridBase_0.4-7 affyio_1.58.0 ellipsis_0.3.1 biomformat_1.16.0
[61] RColorBrewer_1.1-2 hash_2.2.6.1 Rcpp_1.0.5 plyr_1.8.6 progress_1.2.2
[66] zlibbioc_1.34.0 RCurl_1.98-1.2 prettyunits_1.1.1 rpart_4.1-15 openssl_1.4.3
[71] cowplot_1.1.0 zoo_1.8-8 haven_2.3.1 ggrepel_0.8.2 cluster_2.1.0
[76] fs_1.5.0 tinytex_0.27 magrittr_2.0.1 data.table_1.13.4 reprex_0.3.0
[81] mvtnorm_1.1-1 hms_0.5.3 mime_0.9 xtable_1.8-4 XML_3.99-0.5
[86] emdbook_1.3.12 readxl_1.3.1 compiler_4.0.3 biomaRt_2.44.4 bdsmatrix_1.3-4
[91] KernSmooth_2.23-18 crayon_1.3.4 htmltools_0.5.0 mgcv_1.8-33 later_1.1.0.1
[96] geneplotter_1.66.0 lubridate_1.7.9 DBI_1.1.0 dbplyr_1.4.4 rappdirs_0.3.1
[101] MASS_7.3-53 Matrix_1.3-2 ade4_1.7-16 permute_0.9-5 cli_2.1.0
[106] quadprog_1.5-8 marray_1.66.0 gower_0.2.2 igraph_1.2.6 pkgconfig_2.0.3
[111] GenomicAlignments_1.24.0 numDeriv_2016.8-1.1 recipes_0.1.15 xml2_1.3.2 foreach_1.5.1
[116] PCAtools_2.0.0 annotate_1.66.0 dqrng_0.2.1 multtest_2.44.0 prodlim_2019.11.13
[121] rvest_0.3.6 digest_0.6.27 vegan_2.5-6 tm_0.7-8 cellranger_1.1.0
[126] DelayedMatrixStats_1.10.1 curl_4.3 gtools_3.8.2 shiny_1.5.0 Rsamtools_2.4.0
[131] lifecycle_0.2.0 nlme_3.1-151 jsonlite_1.7.1 Rhdf5lib_1.10.1 askpass_1.1
[136] fansi_0.4.1 pillar_1.4.6 fastmap_1.0.1 httr_1.4.2 survival_3.2-7
[141] treemap_2.4-2 glue_1.4.2 xts_0.12.1 iterators_1.0.13 bit_4.0.4
[146] class_7.3-18 stringi_1.5.3 blob_1.2.1 org.Hs.eg.db_3.11.4 BiocSingular_1.4.0
[151] caTools_1.18.0 memoise_1.1.0 irlba_2.3.3 ape_5.4-1
Any advice would be very much appreciated, Thanks !


Hello, Thanks a lot for your answers. It makes sense that depending on the design my assumptions of the calculation of log2FoldChange are more or less wrong. However, even using the simplest possible design for instance
I get very high log2FoldChange for most of DEGs (comparing to what seems biologically relevant to me). If I try to recalculate a simple log2FoldChange as mention before (ok it is bad but just to see), the difference is still very important. With a design as simple as this one, every counts of samples in condition Mortalite="agonie" should be compared to every samples in condition Mortalite="ok" right ? So there should'nt be such a huge gap between values don't you think ?
The "true_logchange" (which isn't true obviously) refers to the calculation as explained before. Here is also my coldata content :
I can also give you a sample of the counts data (example for gene TRINITY_DN1062_c0_g2_i10 shown above) :
So this gene is almost not found in samples in condition "agonie" whereas there are some strong values in samples corresponding to "ok" which explains these high values. Still, for a change in expression from 0.5 average to 120 average, I would expect of log2FoldChange between -5 and -10, not -22 ! So do you think everything is fine here and the "issue" just comes from genes that are not found in some conditions and found in others causing these tremendous changes in expression ?
Should I exclude these genes ?
Again, thanks for the help
H
How do you have half counts? A ratio of zero to anything is going to be really large.
As for excluding them...there is a difference in the means. The software is flagging that for you like it should.. It's up to you if you think that the higher expression levels in a few samples are artifacts that should be ignored. It's not the software's job to make that call for you.
The values returned by
plotCountshave a pseudocount added while the GLM does not use pseudocountsAgain there isn't a bug here.
If you want stable LFCs to use for gene ranking, please use our dedicated function:
lfcShrink. We do think that LFC is important and have spent years working on reliable LFC estimation, but thelogFoldChangecolumn in the MLE from the GLM will tend to infinity if you put in data with all 0's for one of the conditions.Thanks for these useful advices and your reactivity. I now understand better what is going on here.