
Hi, I have MSe data acquired in profile (continuum) mode with a Waters Xevo G2 XS QToF instrument, converted to mzML with MSConvert from proteowizard, and imported using the readMSData(x, centroided = FALSE) function.
I then centroid the data with the pickPeaks function, after smooth and mz-refinement (see code below). My peaks are about 5-10 s wide, and I set the halfWindowSize in smooth() to 2. When trying to save a mzML file (I need this file for other downstream analyses) with writeMSdata I get the error that the halfWindowSize is too big.
data_centroid <- data_profile %>%
smooth(method = "SavitzkyGolay", halfWindowSize = 2L) %>%
pickPeaks(refineMZ = "descendPeak", signalPercentage = 30, msLevel = 1L, SNR = 3, method = "MAD") %>%
filterMsLevel(msLevel=1)
writeMSData(data_centroid, "MSe_RCentroid.mzML", outformat = "mzml")
#Output:
Error: BiocParallel errors
1 remote errors, element index: 1
0 unevaluated and other errors
first remote error:
Error: fun(object@intensity, halfWindowSize = halfWindowSize, ...) : ‘halfWindowSize’ is too large!
When I extract a specific ion, I see that the profile data has many data points per peak.
mz_M4 <- 471.72
mzr <- c(mz_M4 - 0.1, mz_M4 + 0.1)
rtr <- c(240,330)
M4 <- data_profile %>%
filterRt(rtr) %>%
filterMz(mzr)
plot(M4, type = "XIC")
abline(h = mz_M4, col = "red", lty = 2)
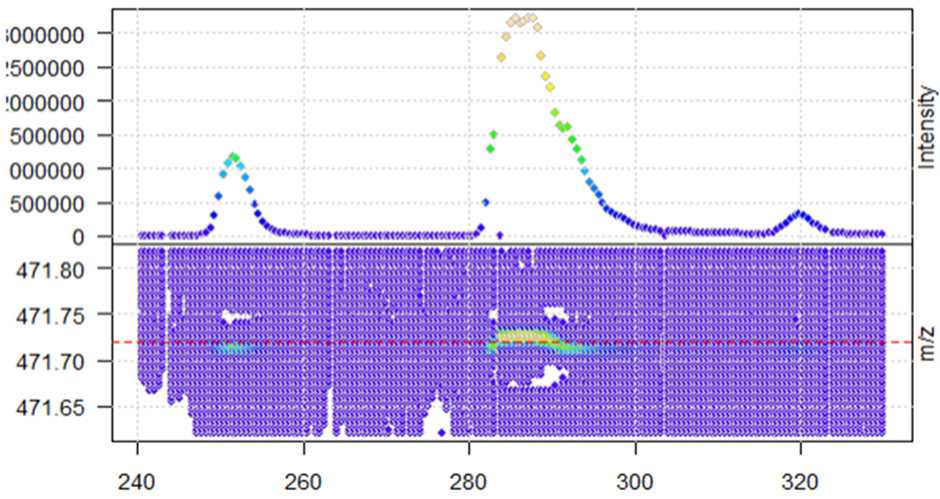
After centroiding with the pickPeaks function above, the function seems to work when looking at the same XIC in the new centroided data (reduction of noise)
M4_cent <- data_centroid %>%
filterRt(rtr) %>%
filterMz(mzr)
plot(M4_cent, type = "XIC")
abline(h = mz_M4, col = "red", lty = 2)
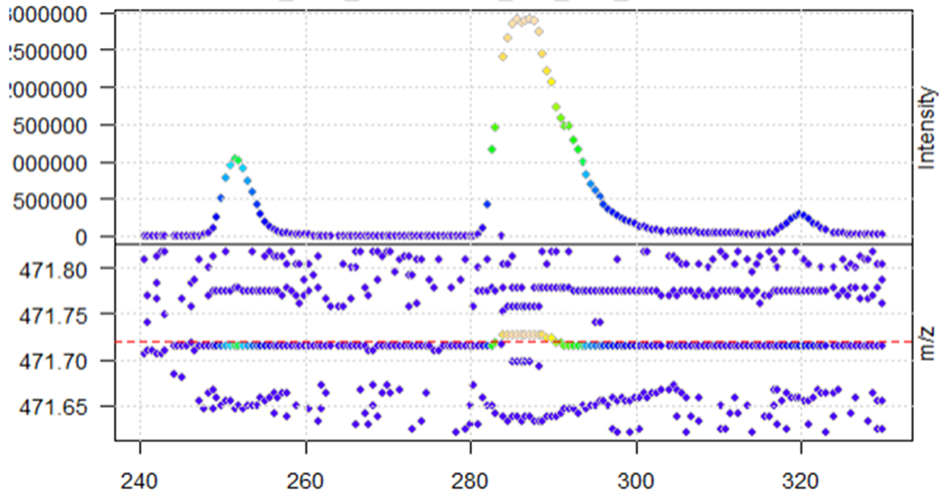
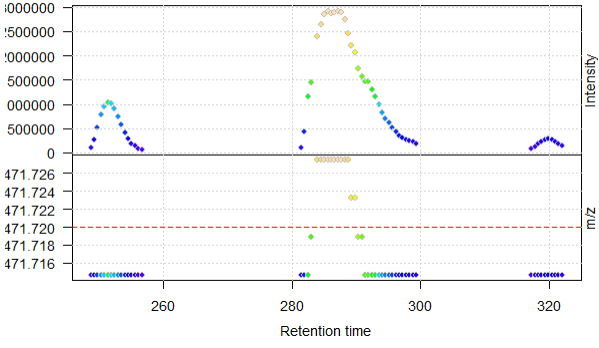
When plotting, I get the warning message that negative intensities were generated
Warning: No data points between 471.62 and 471.82 for spectrum with acquisition number 901. Returning empty spectrum.Warning: No data points between 471.62 and 471.82 for spectrum with acquisition number 976. Returning empty spectrum.Warning: No data points between 471.62 and 471.82 for spectrum with acquisition number 1201. Returning empty spectrum.Warning: Negative intensities generated. Replaced by zeros.Warning: Negative intensities generated. Replaced by zeros.Warning: Negative intensities generated. Replaced by zeros.Warning: Negative intensities generated. Replaced by zeros.Warning: Negative intensities generated. Replaced by zeros.Warning: Negative intensities generated. Replaced by zeros.Warning: Negative intensities generated. Replaced by zeros.Warning: No data points between 471.62 and 471.82 for spectrum with acquisition number 901. Returning empty spectrum.Warning: Negative intensities generated. Replaced by zeros.Warning: Negative intensities generated. Replaced by zeros.Warning: Negative intensities generated. etc
However, these warnings are still there when I use removePeaks() and clean(), which from what I have understood it, should set intensities below the set threshold to zero (removePeaks) and then remove the zero-values (clean), but they seem to be removed in the XIC-plot.
data_centroid <- data_profile %>%
smooth(method = "SavitzkyGolay", halfWindowSize = 2L) %>%
removePeaks(t=50000, verbose = F) %>%
clean(verbose=F, all = T) %>%
pickPeaks(refineMZ = "descendPeak", signalPercentage = 30, msLevel = 1L, SNR = 3, method = "MAD") %>%
filterMsLevel(msLevel=1)
The data can still not be saved with writeMSData().
When I reduce the halfWindowSize to 1 in smooth(), I get a new error
data_centroid <- data_profile %>%
smooth(method = "SavitzkyGolay", halfWindowSize = 1L) %>%
removePeaks(t=50000, verbose = F) %>%
clean(verbose=F, all = T) %>%
pickPeaks(refineMZ = "descendPeak", signalPercentage = 30, msLevel = 1L, SNR = 3, method = "MAD") %>%
filterMsLevel(msLevel=1)
writeMSData(data_centroid, "MSe_RCentroid.mzML", outformat = "mzml")
#Output:
Error: BiocParallel errors
1 remote errors, element index: 1
0 unevaluated and other errors
first remote error:
Error in solve.default(t(X) %*% X): system is computationally singular: reciprocal condition number = 1.19379e-18
I have also tried to remove the smooth() function, but then I got the same error "Error in solve.default(t(X) %*% X): system is computationally singular: reciprocal condition number = 1.19379e-18"
Does anyone have any ideas of how this can be fixed so that I can save the centroided MSe data?
Thanks!
sessionInfo( )
R version 4.2.2 (2022-10-31 ucrt)
Platform: x86_64-w64-mingw32/x64 (64-bit)
Running under: Windows 10 x64 (build 19045)
Matrix products: default
locale:
[1] LC_COLLATE=English_Sweden.utf8 LC_CTYPE=English_Sweden.utf8 LC_MONETARY=English_Sweden.utf8 LC_NUMERIC=C
[5] LC_TIME=English_Sweden.utf8
attached base packages:
[1] stats4 stats graphics grDevices utils datasets methods base
other attached packages:
[1] magrittr_2.0.3 MSnbase_2.24.2 ProtGenerics_1.30.0 S4Vectors_0.36.1 mzR_2.32.0 Rcpp_1.0.9
[7] Biobase_2.58.0 BiocGenerics_0.44.0
loaded via a namespace (and not attached):
[1] tidyselect_1.2.0 xfun_0.35 lattice_0.20-45 colorspace_2.0-3 vctrs_0.5.0 generics_0.1.3
[7] htmltools_0.5.3 yaml_2.3.6 vsn_3.66.0 utf8_1.2.2 XML_3.99-0.13 rlang_1.0.6
[13] pillar_1.8.1 glue_1.6.2 DBI_1.1.3 BiocParallel_1.32.5 affy_1.76.0 foreach_1.5.2
[19] affyio_1.68.0 lifecycle_1.0.3 plyr_1.8.8 mzID_1.36.0 zlibbioc_1.44.0 munsell_0.5.0
[25] pcaMethods_1.90.0 gtable_0.3.1 codetools_0.2-18 evaluate_0.18 knitr_1.41 IRanges_2.32.0
[31] fastmap_1.1.0 doParallel_1.0.17 parallel_4.2.2 fansi_1.0.3 preprocessCore_1.60.2 BiocManager_1.30.19
[37] scales_1.2.1 limma_3.54.0 MsCoreUtils_1.10.0 impute_1.72.3 ggplot2_3.4.0 digest_0.6.30
[43] dplyr_1.0.10 ncdf4_1.21 grid_4.2.2 clue_0.3-63 cli_3.4.1 tools_4.2.2
[49] tibble_3.1.8 cluster_2.1.4 pkgconfig_2.0.3 MASS_7.3-58.1 iterators_1.0.14 assertthat_0.2.1
[55] rmarkdown_2.18 rstudioapi_0.14 R6_2.5.1 MALDIquant_1.22 compiler_4.2.2
Modify Chunk OptionsRun Current Chunk