Entering edit mode

Dear Parisa,
The problem is not to do with the file format. The problem is almost
certainly that you are trying to read data files from different GEO
series
that contain different numbers of rows (probes), and read.maimages()
does
not allow you to do that.
As the help page for read.maimages says, "All image analysis files
being
read are assumed to contain data for the same genelist in the same
order."
Does it make sense to combine the different GEO series? Did they all
use
exactly the same Agilent array? If it does make sense, but the data
files
contain data for different sets of probe, then it is up to you read
the
series into R separately, then to make decisions about which probes
can be
matched up across series and which cannot.
Best wishes
Gordon
-------------- original message -----------------
[BioC] File format for single channel analysis of Agilent microarray
data
with Limma?
Parisa Razaz Parisa.Razaz at icr.ac.uk
Sun May 27 20:09:13 CEST 2012
Hi Guido,
Thank you for getting back to me. I am also using data downloaded from
GEO
and have now incorporated your suggestion of "agilent.median" when
using
the read.maimages function. However the problem now appears to be with
loading files from different series (when using the read.maimages
function). Particular combinations of series work and others don't,
with
those that don't giving the same error message as before. I thought
that
this may be a size limit issue, but the combined number of samples for
some of the series that don't work together is smaller at times than
those
that do. Do you have any idea why this might be and how I would get
around
it?
Thanks,
Parisa
________________________________________
From: Hooiveld, Guido [Guido.Hooiveld@wur.nl]
Sent: 23 May 2012 16:52
To: bioconductor at r-project.org; Parisa Razaz
Subject: RE: [BioC] File format for single channel analysis of Agilent
microarray data with Limma?
Hi Parisa,
I also once struggled with reading in some Agilent singe channel
arrays
(that I downloaded from GEO; GSE27784), but for me these line of codes
worked (in particularly note that the 2nd line is different than the
one
that is given on the website you linked to; specifically the statement
source="agilent.median"):
HTH,
Guido
>
> targets <- readTargets("targets_GSE27784.txt", row.names="Name")
> e.raw <- read.maimages(targets$FileName, source="agilent.median",
green.only=TRUE)
Read GSM686624_251486829200_S01_GE1_105_Jan09_1_1.txt
Read GSM686625_251486829201_S01_GE1_105_Jan09_1_2.txt
Read GSM686626_251486829328_S01_GE1_105_Jan09_1_3.txt
Read GSM686627_251486829200_S01_GE1_105_Jan09_1_2.txt
Read GSM686628_251486829200_S01_GE1_105_Jan09_1_4.txt
Read GSM686629_251486829201_S01_GE1_105_Jan09_1_4.txt
Read GSM686630_251486829328_S01_GE1_105_Jan09_1_4.txt
Read GSM686631_251486829328_S01_GE1_105_Jan09_1_1.txt
Read GSM686632_251486829328_S01_GE1_105_Jan09_1_2.txt
Read GSM686633_251486829200_S01_GE1_105_Jan09_1_3.txt
Read GSM686634_251486829201_S01_GE1_105_Jan09_1_3.txt
Read GSM686635_251486829201_S01_GE1_105_Jan09_1_1.txt
>
> #Background correction using normexp + offset
> e.raw2 <- backgroundCorrect(e.raw, method="normexp", offset=50)
Array 1 corrected
Array 2 corrected
Array 3 corrected
Array 4 corrected
Array 5 corrected
Array 6 corrected
Array 7 corrected
Array 8 corrected
Array 9 corrected
Array 10 corrected
Array 11 corrected
Array 12 corrected
>
> # Perform quantile normalization
> expr.data <- normalizeBetweenArrays(e.raw2, method="quantile")
>
> #Use the avereps function to average replicate spots.
> E.avg <- avereps(expr.data, ID=expr.data$genes$ProbeName)
>
>
> # Alternatively, perform background correction using the negative
control probes + quantile normalization
> table(e.raw$genes$ControlType)
-1 0 1
153 43379 1486
> bg.corr <- neqc(e.raw, status=e.raw$genes$ControlType, negctrl=-1,
regular=0)
>
> E.avg <- avereps(bg.corr, ID=bg.corr$genes$ProbeName)
>
---------------------------------------------------------
Guido Hooiveld, PhD
Nutrition, Metabolism & Genomics Group
Division of Human Nutrition
Wageningen University
Biotechnion, Bomenweg 2
NL-6703 HD Wageningen
the Netherlands
tel: (+)31 317 485788
fax: (+)31 317 483342
email: guido.hooiveld at wur.nl
internet: http://nutrigene.4t.com
http://scholar.google.com/citations?user=qFHaMnoAAAAJ
http://www.researcherid.com/rid/F-4912-2010
-----Original Message-----
From: bioconductor-bounces@r-project.org [mailto:bioconductor-bounces
at r-project.org] On Behalf Of Parisa [guest]
Sent: Wednesday, May 23, 2012 15:51
To: bioconductor at r-project.org; parisa.razaz at icr.ac.uk
Subject: [BioC] File format for single channel analysis of Agilent
microarray data with Limma?
Hi,
I am following the protocol outlined here for analysis of single
channel
Agilent microarray data:
http://matticklab.com/index.php?title=Single_channel_analysis_of_Agile
nt_microarray_data_with_Limma
I keep getting the following error message when using Limma's
read.maimages function to load my data into an RGList object:
Error in RG[[a]][, i] <- obj[, columns[[a]]] :
number of items to replace is not a multiple of replacement length
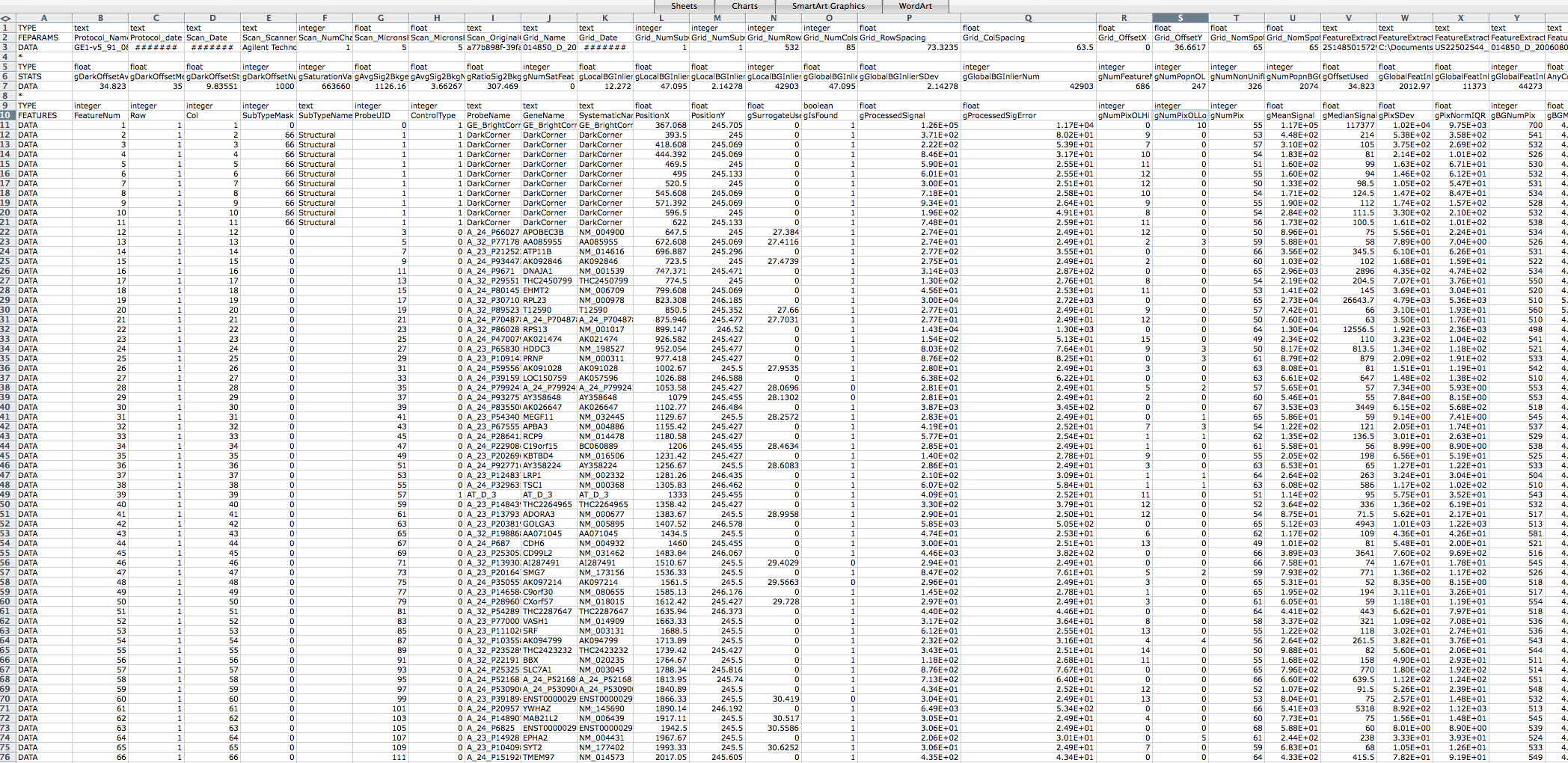
I think this may be due to my Agilent raw data txt files being in the
wrong format. I am having difficulty finding an example Agilent
feature
extraction raw data txt file online to compare it to. A link to a
screen
shot of one of the files I am using is below. I would appreciate if
someone could let me know if it is in the correct format, and if not
then
what format it should be in to prevent the above error message from
coming
up.
Thank you,
Parisa
http://www4.picturepush.com/photo/a/8322602/img/8322602.png
-- output of sessionInfo():
> sessionInfo()R version 2.13.1 (2011-07-08)
Platform: x86_64-apple-darwin9.8.0/x86_64 (64-bit)
locale:
[1] en_GB.UTF-8/en_GB.UTF-8/C/C/en_GB.UTF-8/en_GB.UTF-8
attached base packages:
[1] stats graphics grDevices utils datasets methods base
other attached packages:
[1] limma_3.8.3
______________________________________________________________________
The information in this email is confidential and
intend...{{dropped:4}}

{kind=link}