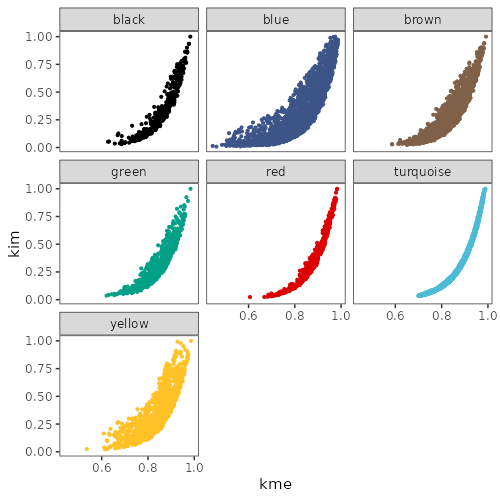
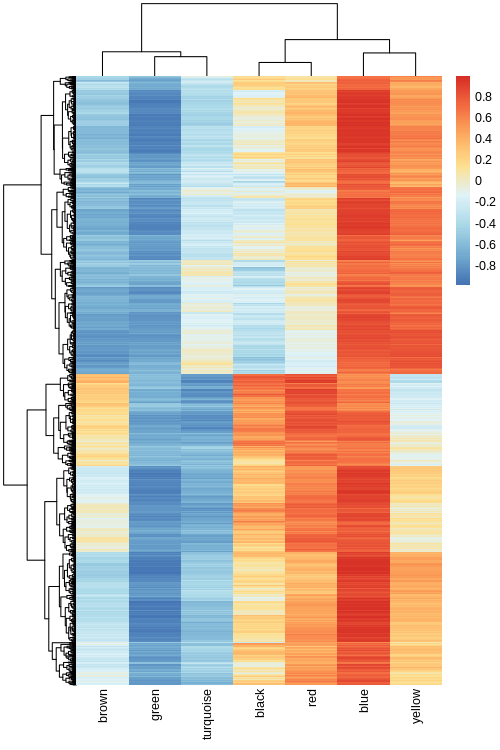
Hello, I've reviewed A: WGCNA Hub Gene Selection Method. I'm calculating module membership (MM) using geneModuleMembership = as.data.frame(cor(datExpr, MEs, use = "p"))
Because kME is a more sophisticated approach as it includes error checking and other features, I also used signedKME( datExpr, MEs, outputColumnName="KME", corFnc="bicor")
For this I'm using reads from the affy hgu133plus2 platform. As a test, I examined the three probes that target a certain gene, and found that the most significant MM correlation was to the tan module in three cases , whereas the kME significance was tan, tan, and dark orange.
However, the dendrogram tree cutting places these three probes in green, gray, and tan. Which of these calculations should I use as the final call for which module a probe most significantly resides within?
Also, kIM only calculates the connectivity of a probe within its home "dendrogram" module, so I'm not easily able to compare kIM values within all the modules? Dr. Langfelder writes kME and kIM are usually very similar, but I have no way to establish this?
Thanks much,
Robert Robl