
I am performing transcript quantification with Salmon, with subsequent differential expression analysis with DESeq2.
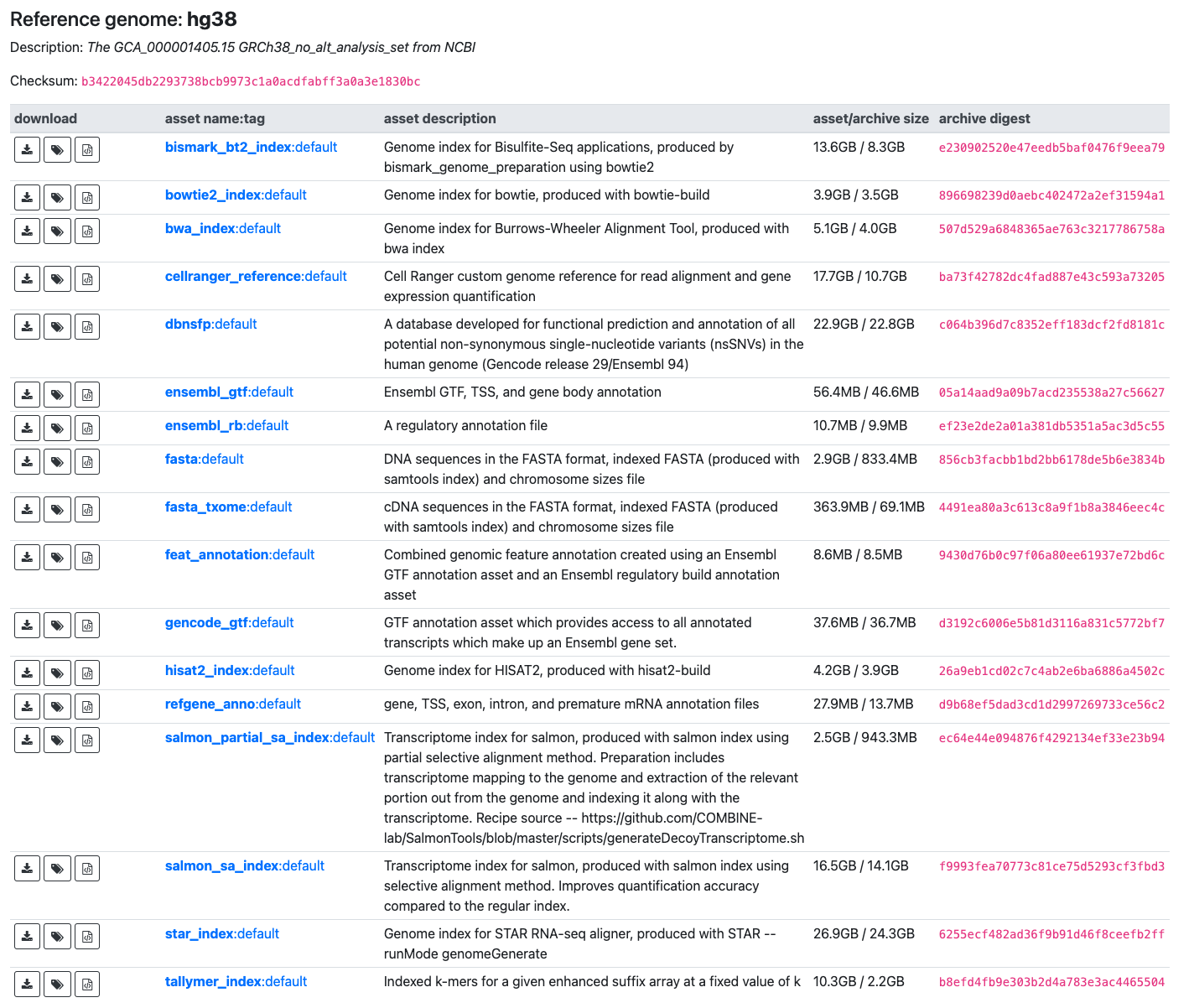
In accordance with the Salmon documentation (https://salmon.readthedocs.io/en/latest/salmon.html), I utilized a pre-built salmon transcriptome index, which I downloaded from refgenie (hg38/salmon_sa_index) - http://refgenomes.databio.org/ (also see screenshot).
Now, my question is as follows: when I import transcript-level estimates with tximport, should I use the TxDb.Hsapiens.UCSC.hg38.knownGene package or the EnsDb.Hsapiens.v86 package to make the tx2gene argument?
Given that the description on refgenie for the hg38 genome is as follows - "The GCA_000001405.15 GRCh38_no_alt_analysis_set from NCBI" (see screenshot), I assume the transcriptome I used was based on USCS annotation, so I assume I should use TxDb.Hsapiens.UCSC.hg38.knownGene. Is that correct?
library(EnsDb.Hsapiens.v86)
edb = EnsDb.Hsapiens.v86
tx = as.data.frame(transcripts(edb, columns = c("tx_name", "gene_id", "gene_name"), return.type="DataFrame"))
tx2gene = tx[, c(1,2)]
#OR#
library(TxDb.Hsapiens.UCSC.hg38.knownGene)
txdb = TxDb.Hsapiens.UCSC.hg38.knownGene
k = keys(txdb, keytype = "TXNAME")
tx2gene = select(txdb, k, "GENEID", "TXNAME")
# library(tximport)
# txi = tximport(files, type = "salmon", tx2gene = tx2gene, ignoreTxVersion=T)
Thank you!

Fantastic, thank you, it's very convenient! Per tximeta output, the matching transcriptome was Ensembl - Homo sapiens - release 97.