
I have gene expression data from some symbiont critters in response to temperature stress, where we replicated temperature treatments across two types of experiments (specifically, in culture and in symbiosis). These separate experiments were both sequenced using TagSeq, but were sequenced at different facilities at different times. The result of both the distinct experiments and using different sequencing facilities is large size factor differences across the two experiments. See here:
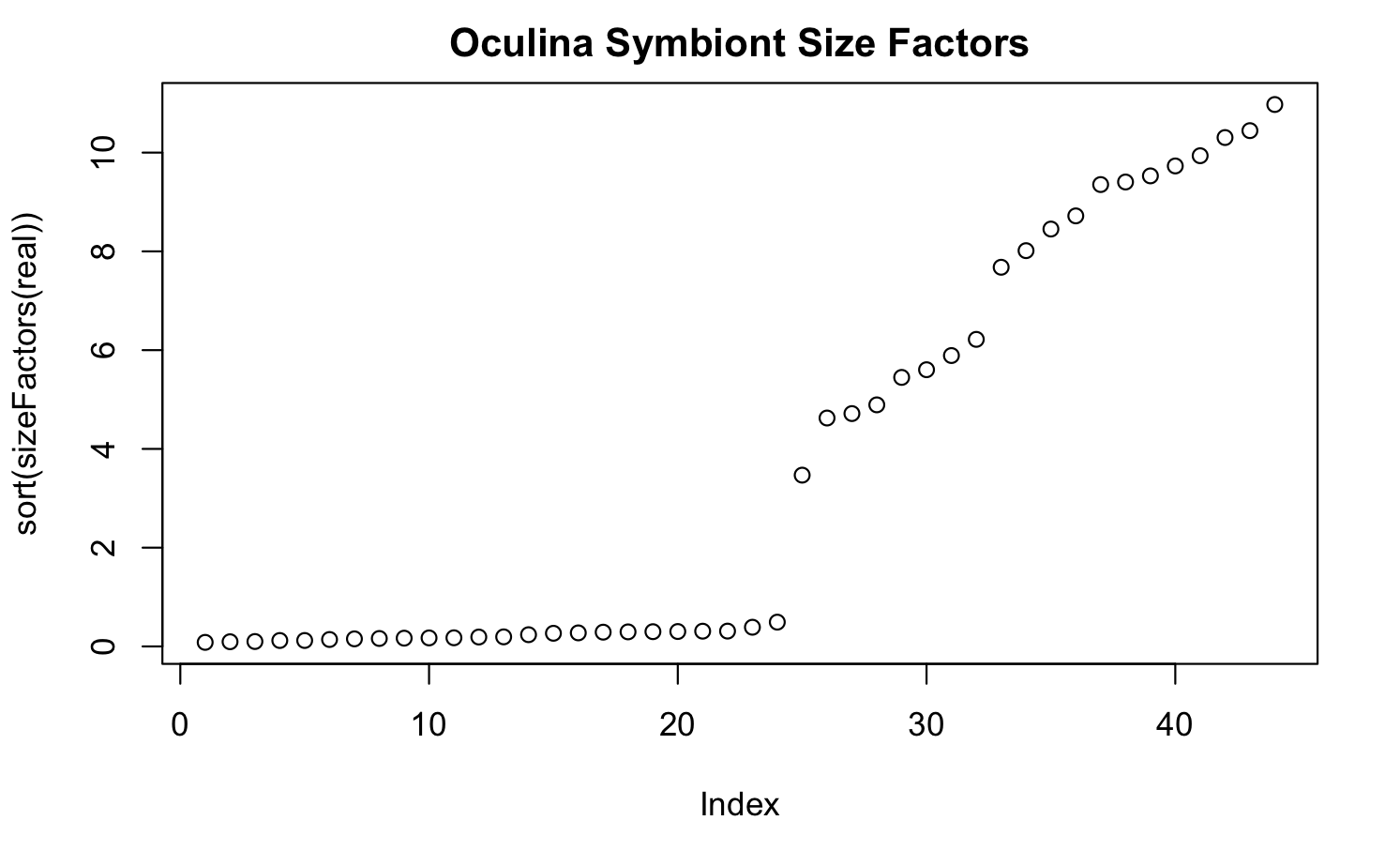
My current course of action for analyzing this data is as follows: raw counts -> batch correction for experiment type using ComBat-seq to get modified counts -> differential expression using DESeq2 to get dds -> rlog transformation -> PCAs.
My question is whether an rlog transformation is appropriate here, specifically considering the different size factors across samples. Additionally, should I be using a blind = FALSE argument in the rlog transformation to account for these size factor differences, since they are associated with the experimental design?
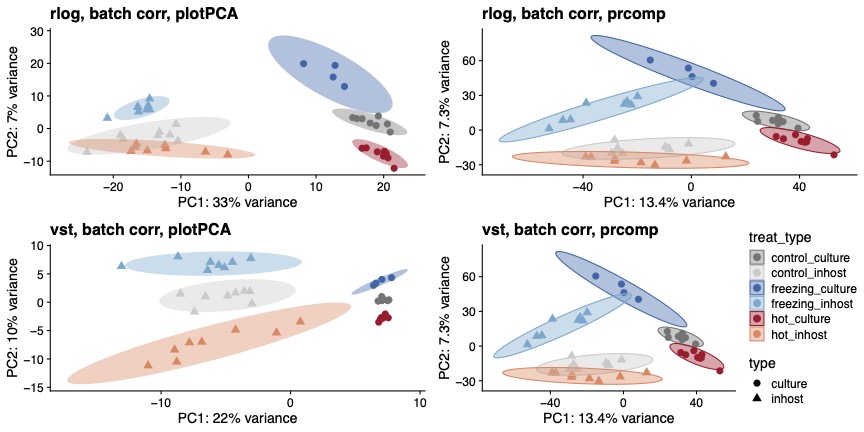
You can see here that, regardless of whether I use vst or rlog, or plotPCA or prcomp, we get separation by experiment type and by temperature treatment. However, I just want to make sure that I am most appropriately making the comparison, since these data are from two separate experiments.

Ok great, thank you so much for your input Michael!