
Hi there, I'm trying to run a differential abundance analyses for my microbiome sequencing data from a multifactorial field trial which looks like this:
- My data is replicated in 'blocks' (bl) that I used as random factor in mixed-effect models (I assume equivalent to 'batch effects' in RNA-seq).
- Within these blocks, I have my nested treatment factors: fertiliser (ft) is nested within variety (vr) which is nested in crop protection (cp).
- based on this experimental design, I recorded the following design formular:
mm = model.matrix(~bl+cp*vr*ft+cp:vr+cp:ft+vr:ft+cp:vr:ft, colData(glomero.dds))
glomero.dds = phyloseq_to_deseq2(ps.filter.glom, design = mm) #ps.filter.glom is a phyloseq object with an otu_table that contains 0s. Depending if I add a pseudo-count or not, I get different results (e.g. if I add 1, it won't run DESeq-function because no overdisersion)
glomero.dds = phyloseq_to_deseq2(ps.filter.glom, design = mm)
gm_mean = function(x, na.rm=TRUE){
exp(sum(log(x[x > 0]), na.rm=na.rm) / length(x))
}
geoMeans = apply(counts(glomero.dds), 1, gm_mean)
glomero.dds = estimateSizeFactors(glomero.dds, geoMeans = geoMeans, type = "poscount") # to be honest, I haven't figured out yet what all these lines with gmMeans do
glomero.dds = DESeq(glomero.dds, test="Wald", fitType="mean")
I ran a PERMANOVA with the adonis function (vegan package) based on a euclidean distance matrix:
deseq_counts_vst.glom <- varianceStabilizingTransformation(glomero.dds)
plotPCA(deseq_counts_vst.glom)
vst_trans_count_tab_full.glom <- assay(deseq_counts_vst.glom)
samp.dist.glom <- dist(t(vst_trans_count_tab_full.glom), method = "euclidian")
adonis(data = meta_field, samp.dist.glom~cp*vr*ft, method = "bray", strata = meta_field$bl) # meta_field is the metadata file from my phyloseq object, strata adds 'block' (bl) as random factor
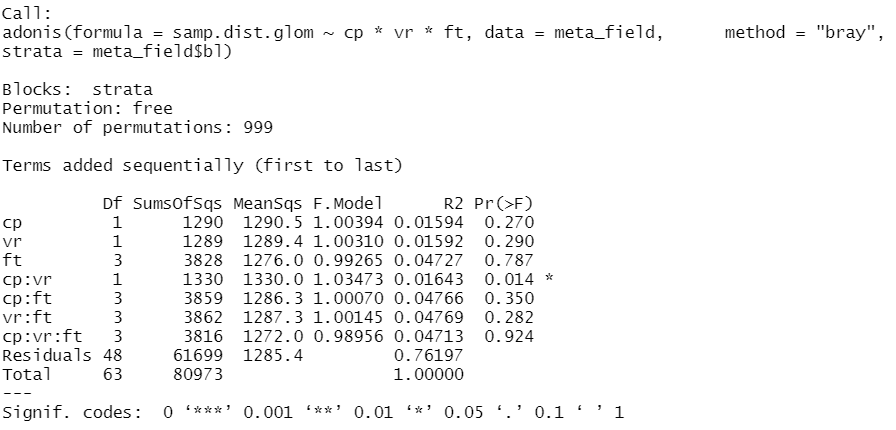
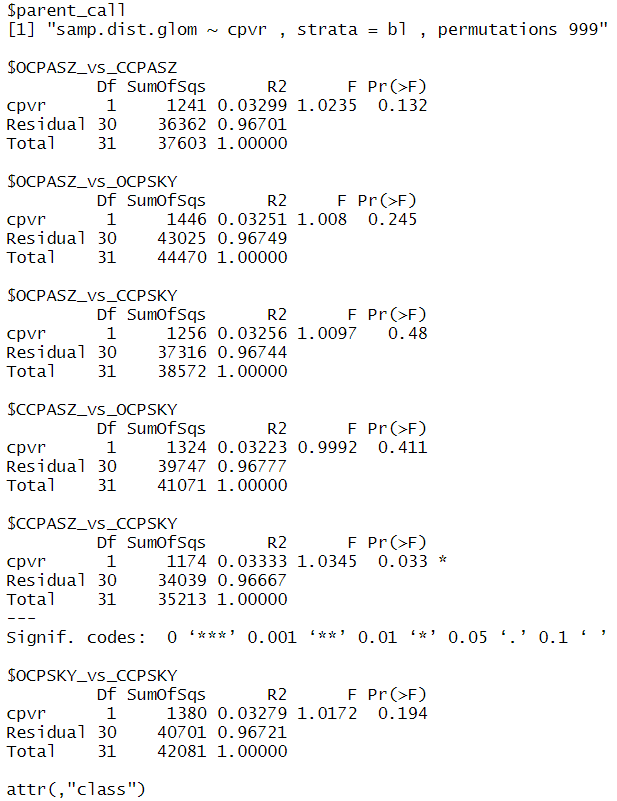
and found out that I have a significant different microbial composition (p = 0.014) with the interaction of variety x crop protection, and specifically, in the comparison of the treatments CCPASZ vs. CCPSKY (CCP means conventional crop protection, ASZ and SKY are the two varieties) which I found out with pairwise.adonis()
pairwise.adonis2(samp.dist.glom ~ cpvr, data = meta_field, strata = 'bl')
Now my problem: the results-function in DESeq2 does not show me any differences that I found with adonis. But when I change the formular, I get different results. However, I cannot specify the contrasts in my result()-function for my cp x vr interaction. When I enter
res.A.B = results(glomero.dds, cooksCutoff = FALSE, contrast = list("cpvr","CCPASZ","CCPSKYZ")) # cpvr is the factor that I used in my meta-data file as a fused factor of cp and vr
I get 'Error in results(glomero.dds, cooksCutoff = FALSE, contrast = c("cpvr", : only list- and numeric-type contrasts are supported for user-supplied model matrices'
When I try to use the interaction from resultsNames(), I get a list with all the contrast that DESeq produces:
matrix(resultsNames(glomero.dds))
[1,] "Intercept"
[2,] "bl2"
[3,] "bl3"
[4,] "bl4"
[5,] "cpOCP"
[6,] "vrSKY"
[7,] "ftCM"
[8,] "ftMN"
[9,] "ftZE"
[10,] "cpOCP.vrSKY"
[11,] "cpOCP.ftCM"
[12,] "cpOCP.ftMN"
[13,] "cpOCP.ftZE"
[14,] "cpCCP.vrSKY.ftCM"
[15,] "cpOCP.vrSKY.ftCM"
[16,] "cpCCP.vrSKY.ftMN"
[17,] "cpOCP.vrSKY.ftMN"
[18,] "cpCCP.vrSKY.ftZE"
[19,] "cpOCP.vrSKY.ftZE"
Number 10: [10,] "cpOCP.vrSKY" would be the right contrast of treatments (cp:vr), but somehow I cannot use this contrast, I either get the same error as before or other error messages.
sigtab.A.B = res.res.A.B = results(glomero.dds, cooksCutoff = FALSE, contrast = c("cpOCP.vrSKY","cpCCP.vrASZ","cpCCP.vrSKY"))
Where is my mistake? Perhpas there are simply no differences between treatment groups according to DESeq2, but I have the feeling that there is sth wrong with my design formular. And if someone could let me know how to use contrast in this specific case, I would be very pleased. I'm very sorry this is far aways from a reproducable example, I don't know how to share the whole phyloseq-object here, but I'm happy to do so!
Thanks a lot!
sessionInfo( )
sessionInfo() R version 4.1.0 (2021-05-18) Platform: x86_64-w64-mingw32/x64 (64-bit) Running under: Windows 10 x64 (build 19041)
attached base packages: 1 parallel stats4 stats graphics grDevices utils datasets methods base
other attached packages:
1 pairwiseAdonis_0.4 cluster_2.1.2 plyr_1.8.6
[4] RColorBrewer_1.1-2 seqinr_4.2-8 scales_1.1.1
[7] extrafont_0.17 vegan_2.5-7 lattice_0.20-44
[10] permute_0.9-5 dendextend_1.15.1 kableExtra_1.3.4
[13] biomformat_1.20.0 phangorn_2.7.0 ape_5.5
[16] DECIPHER_2.20.0 RSQLite_2.2.7 Biostrings_2.60.1
[19] XVector_0.32.0 mctoolsr_0.1.1.2 ggpubr_0.4.0
[22] decontam_1.12.0 DESeq2_1.32.0 SummarizedExperiment_1.22.0
[25] Biobase_2.52.0 MatrixGenerics_1.4.0 matrixStats_0.59.0
[28] GenomicRanges_1.44.0 GenomeInfoDb_1.28.0 IRanges_2.26.0
[31] S4Vectors_0.30.0 BiocGenerics_0.38.0 plotly_4.9.4.1
[34] forcats_0.5.1 stringr_1.4.0 dplyr_1.0.7
[37] purrr_0.3.4 readr_1.4.0 tidyr_1.1.3
[40] tibble_3.1.2 ggplot2_3.3.5 tidyverse_1.3.1
[43] phyloseq_1.36.0 devtools_2.4.2 usethis_2.0.1
[46] ANCOMBC_1.2.2 Maaslin2_1.6.0 dada2_1.20.0
[49] Rcpp_1.0.6
```
