
Hello, I have just started to use DESeq2 and I am trying to compare the results obtained with and without applying lfcShrink. These are the two MA plots, one with lfcShrink and one without:
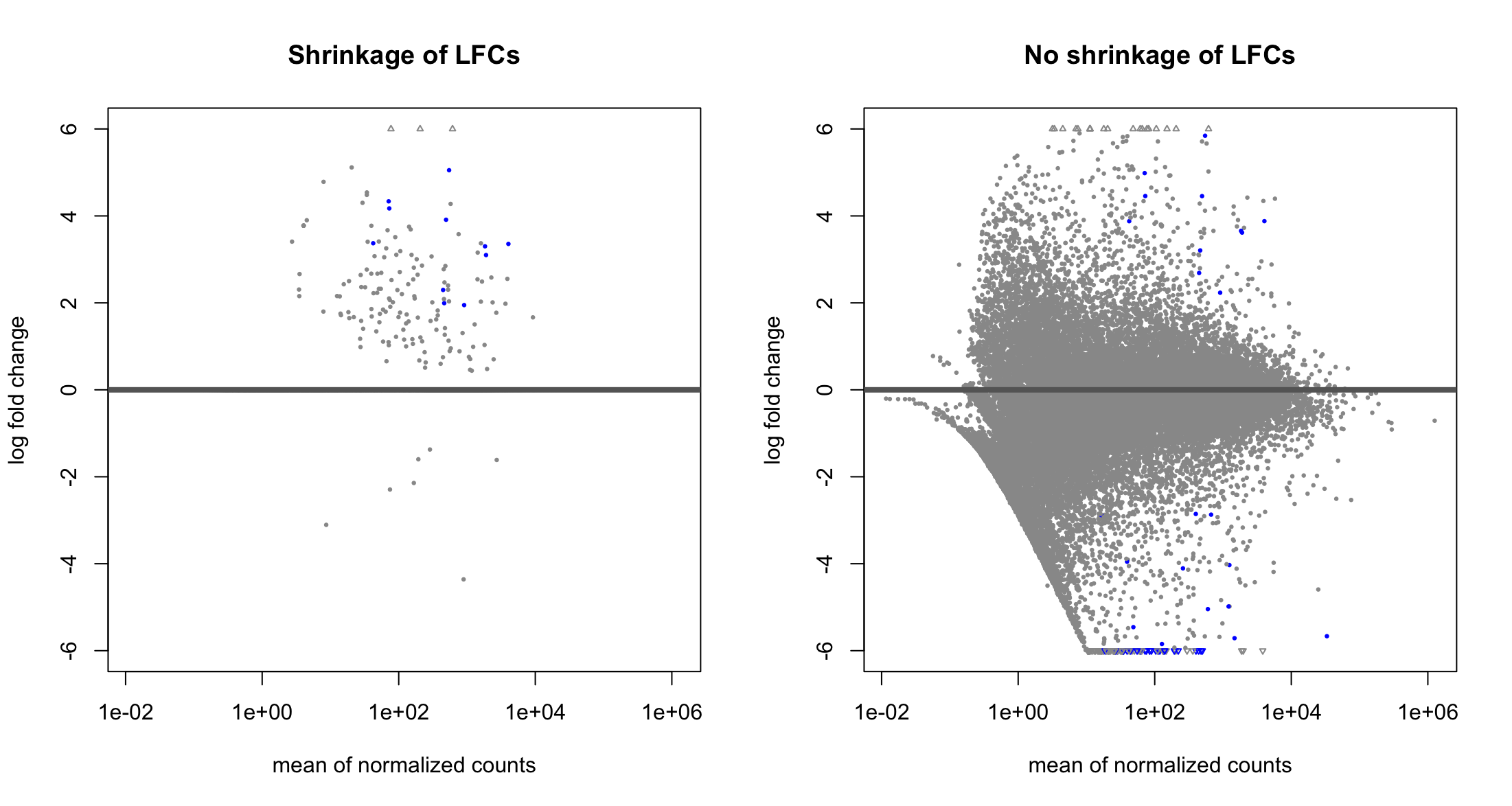
Is it normal to lose all the significant (blue) genes with negative log2FC when applying shrinkage?
I have a very unbalanced dataset, one group with approx. 25-30 samples and the other with 3. But despite this (that I know is not ideal), I would like to understand what is going on in the plots.
Summary of the results with shrinkage:
summary(resLFC)
out of 33014 with nonzero total read count
adjusted p-value < 0.1
LFC > 0 (up) : 15, 0.045%
LFC < 0 (down) : 33, 0.1%
outliers [1] : 586, 1.8%
low counts [2] : 10038, 30%
(mean count < 4)
Summary of the results without shrinkage:
summary(res)
out of 33014 with nonzero total read count
adjusted p-value < 0.1
LFC > 0 (up) : 11, 0.033%
LFC < 0 (down) : 37, 0.11%
outliers [1] : 586, 1.8%
low counts [2] : 10038, 30%
(mean count < 4)
Snippet of the commands (nothing special, just regular commands of DESeq2 afaik):
sampleTable <- data.frame(condition = samples$Responder)
dds <- DESeqDataSetFromTximport(txi, sampleTable, ~condition)
keep <- rowSums(counts(dds)) >= 10
dds <- dds[keep,]
dds = DESeq(dds)
resLFC <- lfcShrink(dds, coef=resultsNames(dds)[2], type="apeglm")
I have also posted the same question in biostars yesterday, but I figured it out that I might be more lucky posting it here :).

Thanks Michael Love ! I am still a bit confused, so then it is normal that all the significant DEG with negative LFC I get without applying
lfcShrink()are gone after applyinglfcShrink()? Because the positive LFC hits I can see that are present in both plots, it is just the negative ones that make me concerned.Yes, well it's just that one method is more conservative at this extreme range of low sample size. Conservative meaning that p-value says the gene is of interest while the lfcShrink method is not convinced.
Another thing to notice: I'm guessing that you are comparing large vs small in your LFC right? So the denominator of the LFC is based on 2 data points? This would explain what is happening.
I see.
Actually, I am comparing the small group (responders) against the big one (non-responders). This is how the 3 MA-plots look now (without shrinkage, with apeglm and with normal). Indeed, using "normal" seems to "improve" it a bit, and I said "improve" because I don't know if it is better or not (probably not easy to assess this).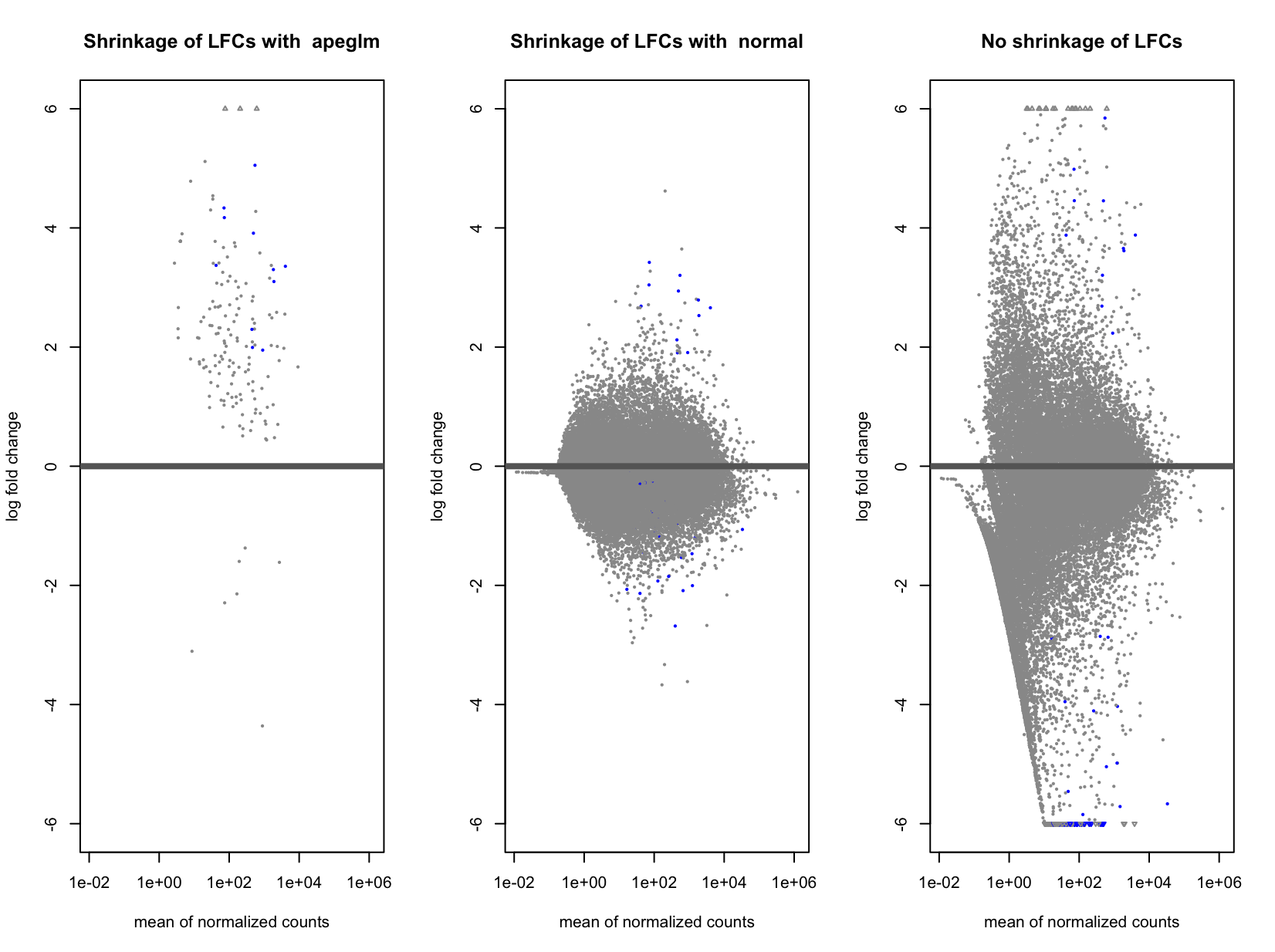
Ok if you are comparing small group (numerator) to big group (denominator) then apeglm is shrinking when you have some counts in the small group but all 0 in the big group. Does the small group have the same sequencing depth as the large group?
Exactly. Sequencing depth is the same yes. I compared a bit the results obtained using the 3 methods:
So without shrinkage, I have highest DEGs, and using apeglm the lowest. And then, all the DEGs in apeglm, are present in normal, and present in no shrinkage.
I think that normal is fine here. apeglm is conservative with very few replicates, which some would find an advantage, but you can just go with "normal".