
Hello, everyone,
I'm recently meeting this problem with my analysis, which i've done a lots of research and asked people around but their answers are quite confusing, so if I can get more opinions, that'd be terrific and thanks at advance.
So I'm doing an analysis of DEGs using Deseq2 and edgeR, the problem here is that I get those two variable, each has two conditions: grippe virus and no, Carcinoma and other. But we really want to figure out is, the impact of virus, but the histological type is quite important as well. So I'm wondering if there is any way that we can skip ou reduce the impact of histological type by designing our module.
It's not an interaction condition, so I built my design code like this:
Code should be placed in three backticks as shown below
DESeq.ds <- DESeqDataSetFromTximport(txi.rsem,sampleTable , ~ condition+Histological_type)
resultsNames(DESeq_new)
[1] "Intercept" "condition_NEG_vs_VIRUS"
[3] "Histological_type_Other_vs_Carcinoma"
DGE.results_histo=results(DESeq_new,pAdjustMethod = "BH",contrast=c("Histological_type","Other","Carcinoma"))
DGE.results_virus=results(DESeq_new,pAdjustMethod = "BH",contrast=c("condition","VIRUS,"NEG"))
So I calculated two conditions, now I've to decide which one should I keep it, Some biologist suggest to find the commun part and get rid of it because what we interested in is virus part. Some of bioinformatist suggest that I need to just build one condition in the first place, like that:
DESeq.ds <- DESeqDataSetFromTximport(txi.rsem,sampleTable , ~ condition)
but in this case I have a lots of outliers cells.
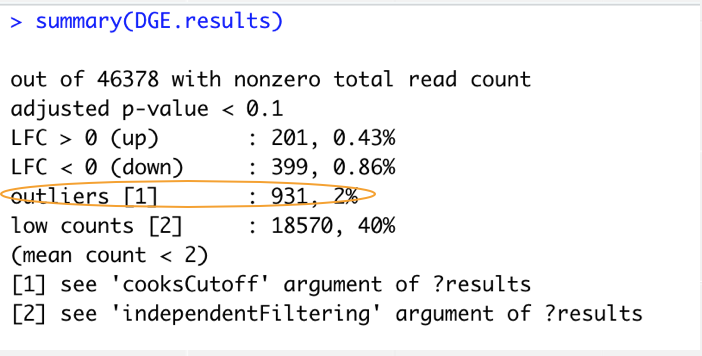
compare to my design with two variables:

I also done the analysis with edgeR, but the part of construct the model makes me less sure, because I don't quite understand what I'm doing, if someone could clarify this part for me would be a great help.
> desigN <- model.matrix(~0+histology_type_edger+Groups_edgeR)
> desigN
histology_type_edgerCarcinoma histology_type_edgerOther Groups_edgeRVIRUS
1 1 0 1
2 1 0 1
3 1 0 1
4 1 0 1
5 0 1 1
6 0 1 1
7 1 0 0
8 1 0 0
9 1 0 0
10 1 0 0
11 0 1 0
12 1 0 0
13 1 0 0
14 1 0 0
15 0 1 0
16 1 0 0
Thanks for your help!!!


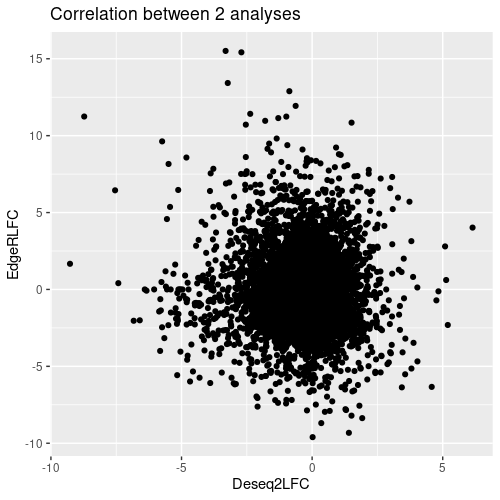
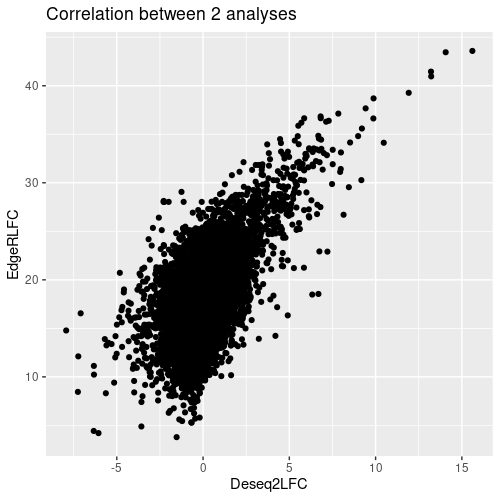
Thanks for your reply, that was what I saw.
But I didn't actually get a very explicable result, so some biologists suggest, that I get rid of the common DEGs of DGE.results_histo and DGE.results_virus from the DEGs of DGE.results_virus, what do you think of this idea?
I don't have much more to add than what I said above.
Thank you for the suggestion, Michael!!