Entering edit mode

Hello,
I am currently running an analysis using DiffBind on Cut&Run data to identify differentially expressed sites. On several occasions, after running DiffBind with default parameters as outlined in the vignette we are getting very low differential peak output from both edgeR and DESeq2 implementations. What could account for not seeing any significantly differentially bound sites?
Below is the output of my command session running DiffBind:
> library(DiffBind)
LTxding required package: GenomicRanges
LTxding required package: stats4
LTxding required package: BiocGenerics
Attaching package: ‘BiocGenerics’
The following objects are masked from ‘package:stats’:
IQR, mad, sd, var, xtabs
The following objects are masked from ‘package:base’:
anyDuplicated, append, as.data.frame, basename, cbind, colnames, dirname, do.call, duplicated,
eval, evalq, Filter, Find, get, grep, grepl, intersect, is.unsorted, lapply, Map, mapply,
match, mget, order, paste, pmax, pmax.int, pmin, pmin.int, Position, rank, rbind, Reduce,
rownames, sapply, setdiff, sort, table, tapply, union, unique, unsplit, which.max, which.min
LTxding required package: S4Vectors
Attaching package: ‘S4Vectors’
The following objects are masked from ‘package:base’:
expand.grid, I, unname
LTxding required package: IRanges
LTxding required package: GenomeInfoDb
LTxding required package: SummarizedExperiment
LTxding required package: MatrixGenerics
LTxding required package: matrixStats
Attaching package: ‘MatrixGenerics’
The following objects are masked from ‘package:matrixStats’:
colAlls, colAnyNAs, colAnys, colAvgsPerRowSet, colCollapse, colCounts, colCummaxs, colCummins,
colCumprods, colCumsums, colDiffs, colIQRDiffs, colIQRs, colLogSumExps, colMadDiffs, colMads,
colMaxs, colMeans2, colMedians, colMins, colOrderStats, colProds, colQuantiles, colRanges,
colRanks, colSdDiffs, colSds, colSums2, colTabulates, colVarDiffs, colVars, colWeightedMads,
colWeightedMeans, colWeightedMedians, colWeightedSds, colWeightedVars, rowAlls, rowAnyNAs,
rowAnys, rowAvgsPerColSet, rowCollapse, rowCounts, rowCummaxs, rowCummins, rowCumprods,
rowCumsums, rowDiffs, rowIQRDiffs, rowIQRs, rowLogSumExps, rowMadDiffs, rowMads, rowMaxs,
rowMeans2, rowMedians, rowMins, rowOrderStats, rowProds, rowQuantiles, rowRanges, rowRanks,
rowSdDiffs, rowSds, rowSums2, rowTabulates, rowVarDiffs, rowVars, rowWeightedMads,
rowWeightedMeans, rowWeightedMedians, rowWeightedSds, rowWeightedVars
LTxding required package: Biobase
Welcome to Bioconductor
Vignettes contain introductory material; view with 'browseVignettes()'. To cite Bioconductor,
see 'citation("Biobase")', and for packages 'citation("pkgname")'.
Attaching package: ‘Biobase’
The following object is masked from ‘package:MatrixGenerics’:
rowMedians
The following objects are masked from ‘package:matrixStats’:
anyMissing, rowMedians
>>> DiffBind 3.4.11
> library(tidyverse)
── Attaching packages ────────────────────────────────────────────────────────────────────── tidyverse 1.3.1 ──
✔ ggplot2 3.3.6 ✔ purrr 0.3.4
✔ tibble 3.1.6 ✔ dplyr 1.0.9
✔ tidyr 1.2.0 ✔ stringr 1.4.0
✔ readr 2.1.2 ✔ forcats 0.5.1
── Conflicts ───────────────────────────────────────────────────────────────────────── tidyverse_conflicts() ──
✖ dplyr::collapse() masks IRanges::collapse()
✖ dplyr::combine() masks Biobase::combine(), BiocGenerics::combine()
✖ dplyr::count() masks matrixStats::count()
✖ dplyr::desc() masks IRanges::desc()
✖ tidyr::expand() masks S4Vectors::expand()
✖ dplyr::filter() masks stats::filter()
✖ dplyr::first() masks S4Vectors::first()
✖ dplyr::lag() masks stats::lag()
✖ ggplot2::Position() masks BiocGenerics::Position(), base::Position()
✖ purrr::reduce() masks GenomicRanges::reduce(), IRanges::reduce()
✖ dplyr::rename() masks S4Vectors::rename()
✖ dplyr::slice() masks IRanges::slice()
> library(rtracklayer)
> #Check column names______________
> names(h3k27me3broad_unique)
[1] "SampleID" "Tissue" "Factor" "Condition" "Treatment" "Replicate" "bamReads" "ControlID"
[9] "bamControl" "Peaks" "PeakCaller"
> #Read in peaksets______________
> h3k27me3broad_unique <- dba(sampleSheet=h3k27me3broad_unique)
veh.rep1 cellLine H3K27me3 vehicle Control 1 narrow
veh.rep2 cellLine H3K27me3 vehicle Control 2 narrow
veh.rep3 cellLine H3K27me3 vehicle Control 3 narrow
Tx.rep1 cellLine H3K27me3 Tx treatment 1 narrow
Tx.rep2 cellLine H3K27me3 Tx treatment 2 narrow
Tx.rep3 cellLine H3K27me3 Tx treatment 3 narrow
> #Check that peaks were identified. # of matrix sites should be > 0 and Intervals column gernerated
> h3k27me3broad_unique
6 Samples, 32872 sites in matrix (98986 total):
ID Tissue Factor Condition Treatment Replicate Intervals
1 veh.rep1 cellLine H3K27me3 vehicle Control 1 19001
2 veh.rep2 cellLine H3K27me3 vehicle Control 2 47836
3 veh.rep3 cellLine H3K27me3 vehicle Control 3 29014
4 Tx.rep1 cellLine H3K27me3 Tx treatment 1 32643
5 Tx.rep2 cellLine H3K27me3 Tx treatment 2 25650
6 Tx.rep3 cellLine H3K27me3 Tx treatment 3 9167
> #Compute count information for each region (Affinity binding matrix):______________
> #bUseSummarizeOverlaps: to use a more standard counting procedure than the built-in one by default.
> h3k27me3broad_unique <- dba.count(h3k27me3broad_unique, bUseSummarizeOverlaps=TRUE)
Computing summits...
Re-centering peaks...
> #FRiP value:______________
> h3k27me3broad_unique
6 Samples, 30833 sites in matrix:
ID Tissue Factor Condition Treatment Replicate Reads FRiP
1 veh.rep1 cellLine H3K27me3 vehicle Control 1 11137455 0.05
2 veh.rep2 cellLine H3K27me3 vehicle Control 2 11515996 0.06
3 veh.rep3 cellLine H3K27me3 vehicle Control 3 10348575 0.06
4 Tx.rep1 cellLine H3K27me3 Tx treatment 1 11752746 0.04
5 Tx.rep2 cellLine H3K27me3 Tx treatment 2 10714697 0.05
6 Tx.rep3 cellLine H3K27me3 Tx treatment 3 6604986 0.05
> #Establish comparisons to make (Contrast):______________
> #categories changed based on which column of metadata will be "compared:
> h3k27me3broad_unique <- dba.contrast(h3k27me3broad_unique,categories=DBA_CONDITION)
Computing results names...
> #Below prevents BPPARAM error during dba.analyze______________
> library(BiocParallel)
> register(SerialParam())
'BiocParallel' did not register default BiocParallelParam:
missing value where TRUE/FALSE needed
> #Differential Enrichment Analysis:______________
> h3k27me3broad_unique <- dba.analyze(h3k27me3broad_unique, method=DBA_ALL_METHODS)
Applying Blacklist/Greylists...
Genome detected: Hsapiens.UCSC.hg38
Applying blacklist...
Removed: 45 of 30833 intervals.
Counting control reads for greylist...
Building greylist: /projects/b1122/Mariana/RAW_data/Cut_and_Run/data/bams/2-NE_S10.bam
coverage: 10942705 bp (0.35%)
Master greylist: 596 ranges, 10942705 bases
Removed: 80 of 30788 intervals.
Removed 125 (of 30833) consensus peaks.
Normalize edgeR with defaults...
Normalize DESeq2 with defaults...
Analyzing...
warning: solve(): system is singular (rcond: 5.44457e-17); attempting approx solution
warning: solve(): system is singular (rcond: 6.74445e-17); attempting approx solution
warning: solve(): system is singular (rcond: 8.3574e-17); attempting approx solution
warning: solve(): system is singular (rcond: 1.22132e-16); attempting approx solution
> #Summary of results for each tool (edgeR often is more stringent --> fewer peaks)______________
> dba.show(h3k27me3broad_unique, bContrasts=T)
Factor Group Samples Group2 Samples2 DB.edgeR DB.DESeq2
1 Condition Tx 3 vehicle 3 0 1
sessionInfo( )
R version 4.1.1 (2021-08-10)
Platform: x86_64-pc-linux-gnu (64-bit)
Running under: Red Hat Enterprise Linux Server 7.5 (Maipo)
Matrix products: default
BLAS: /hpc/software/R/4.1.1/lib64/R/lib/libRblas.so
LAPACK: /hpc/software/R/4.1.1/lib64/R/lib/libRlapack.so
locale:
[1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C LC_TIME=en_US.UTF-8
[4] LC_COLLATE=en_US.UTF-8 LC_MONETARY=en_US.UTF-8 LC_MESSAGES=en_US.UTF-8
[7] LC_PAPER=en_US.UTF-8 LC_NAME=C LC_ADDRESS=C
[10] LC_TELEPHONE=C LC_MEASUREMENT=en_US.UTF-8 LC_IDENTIFICATION=C
attached base packages:
[1] stats4 stats graphics grDevices utils datasets methods base
other attached packages:
[1] Rsubread_2.8.2 mixsqp_0.3-47 rtracklayer_1.54.0
[4] forcats_0.5.1 stringr_1.4.0 dplyr_1.0.9
[7] purrr_0.3.4 readr_2.1.2 tidyr_1.2.0
[10] tibble_3.1.6 ggplot2_3.3.6 tidyverse_1.3.1
[13] DiffBind_3.4.11 SummarizedExperiment_1.24.0 Biobase_2.54.0
[16] MatrixGenerics_1.6.0 matrixStats_0.62.0 GenomicRanges_1.46.1
[19] GenomeInfoDb_1.30.1 IRanges_2.28.0 S4Vectors_0.32.4
[22] BiocGenerics_0.40.0
loaded via a namespace (and not attached):
[1] amap_0.8-18 colorspace_2.0-2 rjson_0.2.21
[4] deldir_1.0-6 hwriter_1.3.2.1 ellipsis_0.3.2
[7] XVector_0.34.0 fs_1.5.2 rstudioapi_0.13
[10] ggrepel_0.9.1 bit64_4.0.5 lubridate_1.8.0
[13] AnnotationDbi_1.56.2 fansi_1.0.2 mvtnorm_1.1-3
[16] apeglm_1.16.0 xml2_1.3.3 splines_4.1.1
[19] cachem_1.0.6 geneplotter_1.72.0 jsonlite_1.8.0
[22] Rsamtools_2.10.0 broom_1.0.0 annotate_1.72.0
[25] dbplyr_2.2.1 ashr_2.2-54 png_0.1-7
[28] GreyListChIP_1.26.0 compiler_4.1.1 httr_1.4.3
[31] backports_1.4.1 assertthat_0.2.1 Matrix_1.3-4
[34] fastmap_1.1.0 limma_3.50.3 cli_3.3.0
[37] htmltools_0.5.2 tools_4.1.1 coda_0.19-4
[40] gtable_0.3.0 glue_1.6.2 GenomeInfoDbData_1.2.7
[43] systemPipeR_2.0.8 ShortRead_1.52.0 Rcpp_1.0.8.3
[46] bbmle_1.0.25 cellranger_1.1.0 vctrs_0.4.1
[49] Biostrings_2.62.0 rvest_1.0.2 lifecycle_1.0.1
[52] irlba_2.3.5 restfulr_0.0.15 gtools_3.9.2.2
[55] XML_3.99-0.10 zlibbioc_1.40.0 MASS_7.3-54
[58] scales_1.1.1 BSgenome_1.62.0 hms_1.1.1
[61] parallel_4.1.1 RColorBrewer_1.1-2 yaml_2.3.5
[64] memoise_2.0.1 emdbook_1.3.12 bdsmatrix_1.3-6
[67] latticeExtra_0.6-30 stringi_1.7.6 RSQLite_2.2.14
[70] SQUAREM_2021.1 genefilter_1.76.0 BiocIO_1.4.0
[73] caTools_1.18.2 BiocParallel_1.28.3 truncnorm_1.0-8
[76] rlang_1.0.3 pkgconfig_2.0.3 bitops_1.0-7
[79] lattice_0.20-44 invgamma_1.1 GenomicAlignments_1.30.0
[82] htmlwidgets_1.5.4 bit_4.0.4 tidyselect_1.1.2
[85] plyr_1.8.7 magrittr_2.0.3 DESeq2_1.34.0
[88] R6_2.5.1 gplots_3.1.3 generics_0.1.2
[91] DelayedArray_0.20.0 DBI_1.1.3 withr_2.5.0
[94] haven_2.5.0 pillar_1.7.0 survival_3.2-11
[97] KEGGREST_1.34.0 RCurl_1.98-1.7 modelr_0.1.8
[100] crayon_1.4.2 interp_1.1-2 KernSmooth_2.23-20
[103] utf8_1.2.2 tzdb_0.3.0 jpeg_0.1-9
[106] readxl_1.4.0 locfit_1.5-9.5 grid_4.1.1
[109] blob_1.2.3 reprex_2.0.1 digest_0.6.29
[112] xtable_1.8-4 numDeriv_2016.8-1.1 munsell_0.5.0

I will look into plots of the data more thoroughly and see if any of the trends fit the reasons for low differential binding sites! Thank you for your insight.
Dr. Stark, thank you for your insight several days ago! I went back to check the quality of our Cut&Run peaks (called with MACS2) using ChIPQC as well as ran a number of the exploratory plots you suggested in the original post. We are specifically interested in looking at the broad peak file generated from MACS2. Additionally, we went back and checked our library qualities as well as bam mapping QC, both of which appeared normal. Below are the results from ChIPQC & exploratory plots:
Results from H3K27 ChIPQC - showing low RiP%. Notably, in our other samples looking at H3K4, RiP% was significantly higher (in the 50%-60% range)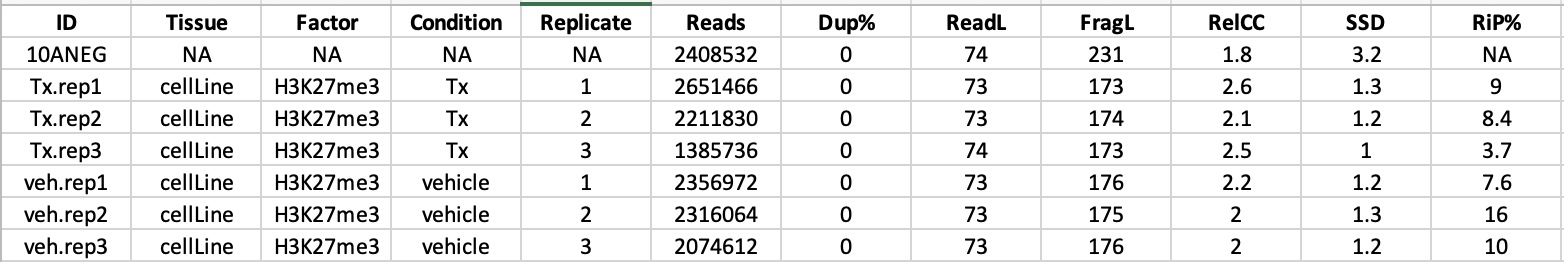
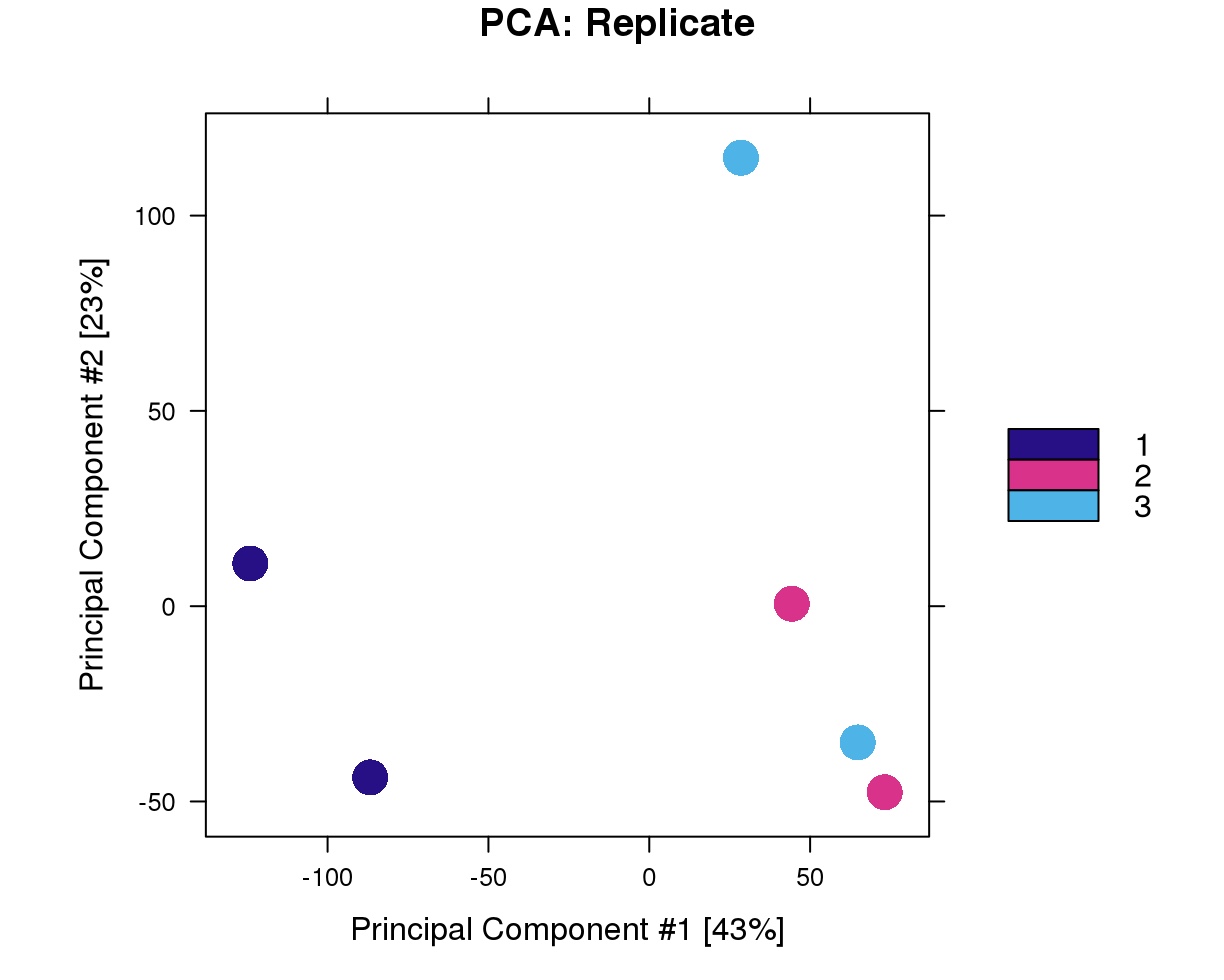
PCA Results based on condition & replicates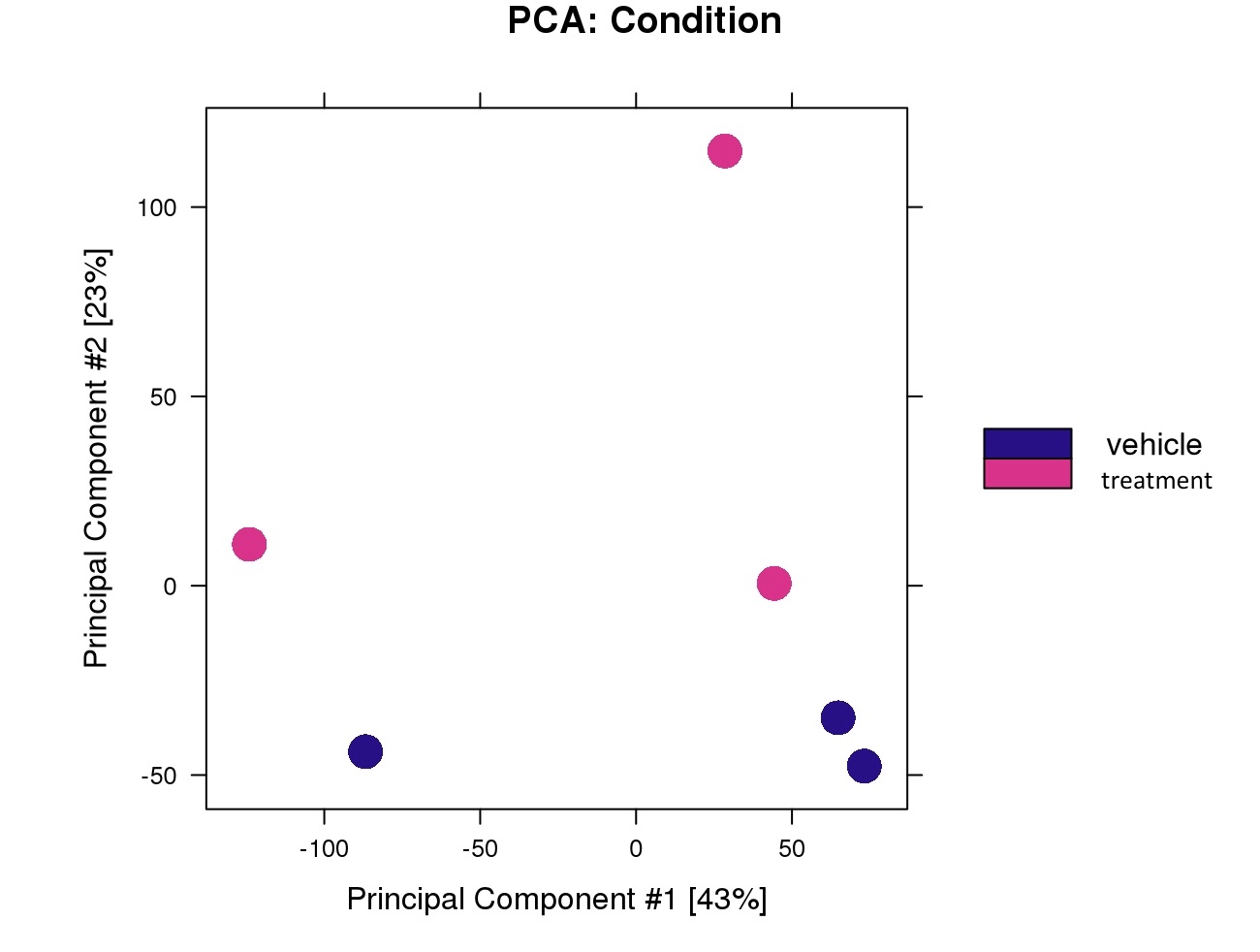
dba.show results after dba.count. Note: I set summits to fit the mean peak size, 442. Using default parameters for dba.count yielded only 1 differential site. Could this be because for H3K27 methylation sites are more broad?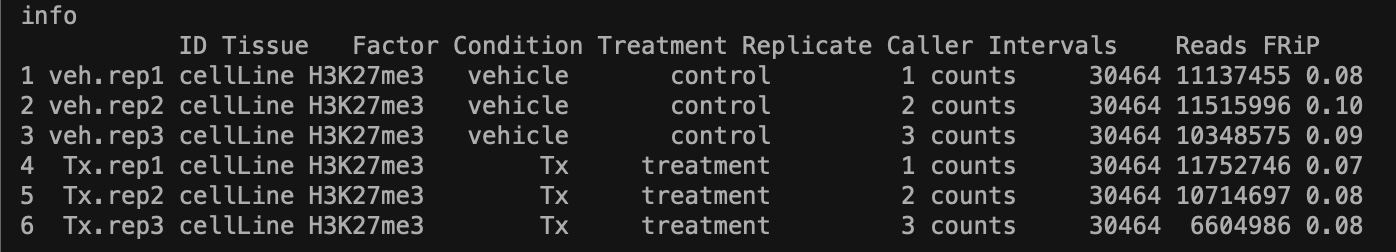
MA plot comparing treatment and control. Based on this plot, how would one determine if the result due to technical variance or biological variance?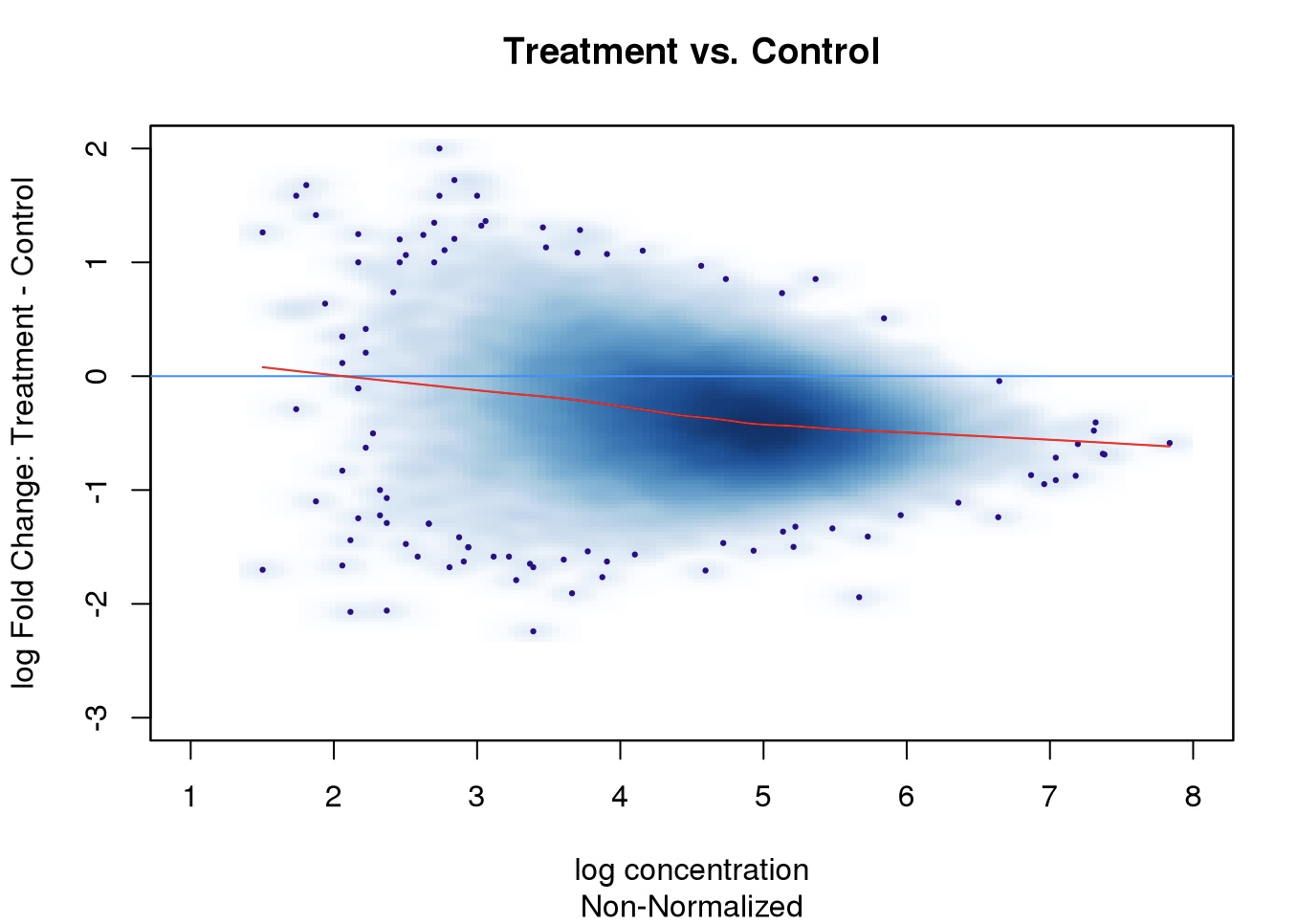
I appreciate any insight you may have and can also provide more information as needed! My session info is attached below. Also, I apologize for having to input data from the command line as an image. The code on the comment box would not format it properly so it would be readable.
R version 4.1.1 (2021-08-10) Platform: x86_64-pc-linux-gnu (64-bit) Running under: Red Hat Enterprise Linux Server 7.5 (Maipo)
Matrix products: default BLAS: /hpc/software/R/4.1.1/lib64/R/lib/libRblas.so LAPACK: /hpc/software/R/4.1.1/lib64/R/lib/libRlapack.so
locale: 1 LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C LC_TIME=en_US.UTF-8 LC_COLLATE=en_US.UTF-8
5 LC_MONETARY=en_US.UTF-8 LC_MESSAGES=en_US.UTF-8 LC_PAPER=en_US.UTF-8 LC_NAME=C
[9] LC_ADDRESS=C LC_TELEPHONE=C LC_MEASUREMENT=en_US.UTF-8 LC_IDENTIFICATION=C
attached base packages: 1 stats4 stats graphics grDevices utils datasets methods base
other attached packages: 1 BSgenome.Hsapiens.UCSC.hg38_1.4.4 BiocManager_1.30.18 GreyListChIP_1.26.0
4 BSgenome_1.62.0 Biostrings_2.62.0 XVector_0.34.0
[7] edgeR_3.36.0 limma_3.50.3 rtracklayer_1.54.0
[10] forcats_0.5.1 stringr_1.4.0 dplyr_1.0.9
[13] purrr_0.3.4 readr_2.1.2 tidyr_1.2.0
[16] tibble_3.1.6 tidyverse_1.3.1 BiocParallel_1.28.3
[19] TxDb.Hsapiens.UCSC.hg38.knownGene_3.14.0 GenomicFeatures_1.46.5 AnnotationDbi_1.56.2
[22] ChIPQC_1.30.0 DiffBind_3.4.11 SummarizedExperiment_1.24.0
[25] Biobase_2.54.0 MatrixGenerics_1.6.0 matrixStats_0.62.0
[28] GenomicRanges_1.46.1 GenomeInfoDb_1.30.1 IRanges_2.28.0
[31] S4Vectors_0.32.4 BiocGenerics_0.40.0 ggplot2_3.3.6
loaded via a namespace (and not attached): 1 readxl_1.4.0 backports_1.4.1
3 BiocFileCache_2.2.1 plyr_1.8.7
5 splines_4.1.1 amap_0.8-18
[7] digest_0.6.29 invgamma_1.1
[9] htmltools_0.5.2 SQUAREM_2021.1
[11] fansi_1.0.2 magrittr_2.0.3
[13] memoise_2.0.1 tzdb_0.3.0
[15] annotate_1.72.0 Nozzle.R1_1.1-1.1
[17] modelr_0.1.8 systemPipeR_2.0.8
[19] bdsmatrix_1.3-6 prettyunits_1.1.1
[21] jpeg_0.1-9 colorspace_2.0-2
[23] rvest_1.0.2 blob_1.2.3
[25] rappdirs_0.3.3 apeglm_1.16.0
[27] ggrepel_0.9.1 haven_2.5.0
[29] jsonlite_1.8.0 crayon_1.4.2
[31] RCurl_1.98-1.7 TxDb.Hsapiens.UCSC.hg19.knownGene_3.2.2
[33] chipseq_1.44.0 genefilter_1.76.0
[35] survival_3.2-11 glue_1.6.2
[37] gtable_0.3.0 zlibbioc_1.40.0
[39] DelayedArray_0.20.0 scales_1.1.1
[41] mvtnorm_1.1-3 DBI_1.1.3
[43] TxDb.Mmusculus.UCSC.mm9.knownGene_3.2.2 Rcpp_1.0.8.3
[45] xtable_1.8-4 progress_1.2.2
[47] emdbook_1.3.12 bit_4.0.4
[49] truncnorm_1.0-8 htmlwidgets_1.5.4
[51] httr_1.4.3 gplots_3.1.3
[53] RColorBrewer_1.1-2 ellipsis_0.3.2
[55] pkgconfig_2.0.3 XML_3.99-0.10
[57] dbplyr_2.2.1 deldir_1.0-6
[59] locfit_1.5-9.5 utf8_1.2.2
[61] tidyselect_1.1.2 rlang_1.0.3
[63] reshape2_1.4.4 cellranger_1.1.0
[65] munsell_0.5.0 tools_4.1.1
[67] cachem_1.0.6 cli_3.3.0
[69] generics_0.1.3 RSQLite_2.2.14
[71] broom_1.0.0 fastmap_1.1.0
[73] yaml_2.3.5 fs_1.5.2
[75] bit64_4.0.5 caTools_1.18.2
[77] KEGGREST_1.34.0 TxDb.Rnorvegicus.UCSC.rn4.ensGene_3.2.2
[79] xml2_1.3.3 biomaRt_2.50.3
[81] compiler_4.1.1 rstudioapi_0.13
[83] filelock_1.0.2 curl_4.3.2
[85] png_0.1-7 reprex_2.0.1
[87] geneplotter_1.72.0 stringi_1.7.6
[89] TxDb.Hsapiens.UCSC.hg18.knownGene_3.2.2 lattice_0.20-44
[91] Matrix_1.3-4 vctrs_0.4.1
[93] pillar_1.7.0 lifecycle_1.0.1
[95] bitops_1.0-7 irlba_2.3.5
[97] TxDb.Mmusculus.UCSC.mm10.knownGene_3.10.0 TxDb.Celegans.UCSC.ce6.ensGene_3.2.2
[99] R6_2.5.1 BiocIO_1.4.0
[101] latticeExtra_0.6-30 hwriter_1.3.2.1
[103] ShortRead_1.52.0 KernSmooth_2.23-20
[105] MASS_7.3-54 gtools_3.9.2.2
[107] assertthat_0.2.1 DESeq2_1.34.0
[109] rjson_0.2.21 withr_2.5.0
[111] GenomicAlignments_1.30.0 Rsamtools_2.10.0
[113] GenomeInfoDbData_1.2.7 parallel_4.1.1
[115] hms_1.1.1 grid_4.1.1
[117] TxDb.Dmelanogaster.UCSC.dm3.ensGene_3.2.2 coda_0.19-4
[119] ashr_2.2-54 mixsqp_0.3-47
[121] bbmle_1.0.25 lubridate_1.8.0
[123] numDeriv_2016.8-1.1 interp_1.1-2
[125] restfulr_0.0.15