
Background: I am comparing 6 colorectal tumor samples to 19 Normal samples using limma-RNASeq. I followed standard steps as suggested in the limma manual (hence not printing the code) to predict differential gene expression in Tumor samples compared to 19 Normals. Tumors: There were six biopsies from a colorectal cancer patient and all of them were sequenced using the same protocols. Normal: These are 19 distinct normal tissue ( i.e only 1 sample per tissue type. 1-heart, 1 lung, 1-Colon,1-skin, 1 testis, and so on) samples from a donor sequenced using the same library protocol. Hence likely no batch effect due to library pre-methods.
Issue: Although for the most part DEG analysis worked, there were some unexpected results. For example, Limma predicted a set of genes that belong to a gene family called "CEACAM[1-5]", which are profoundly expressed in normal intestinal epithelial tissue ( high in the normal colon compared to other tissue types ) as a differentially expressed gene with the highest log-FD change while one of the samples (1/19) in the Normal cohort has a similar expression as 6 tumor samples. But this isn't true as its highly expressed (comparable expression to tumor samples) in the normal Colon ( only one sample in the Normal cohort). There are multiple other instances where limma falsely predicts "outliers" tissue-specific genes as Differentially expressed genes.
Actual question: What are the options/ways to handle such "outlier gene" cases in limma. ? Options: Is that happening because I have only one colon sample in my normal cohort? IF yes, what are the recommended number of samples of each type needed to rectify these issues?
Data: Following is TMM normalized log2(TPM) values
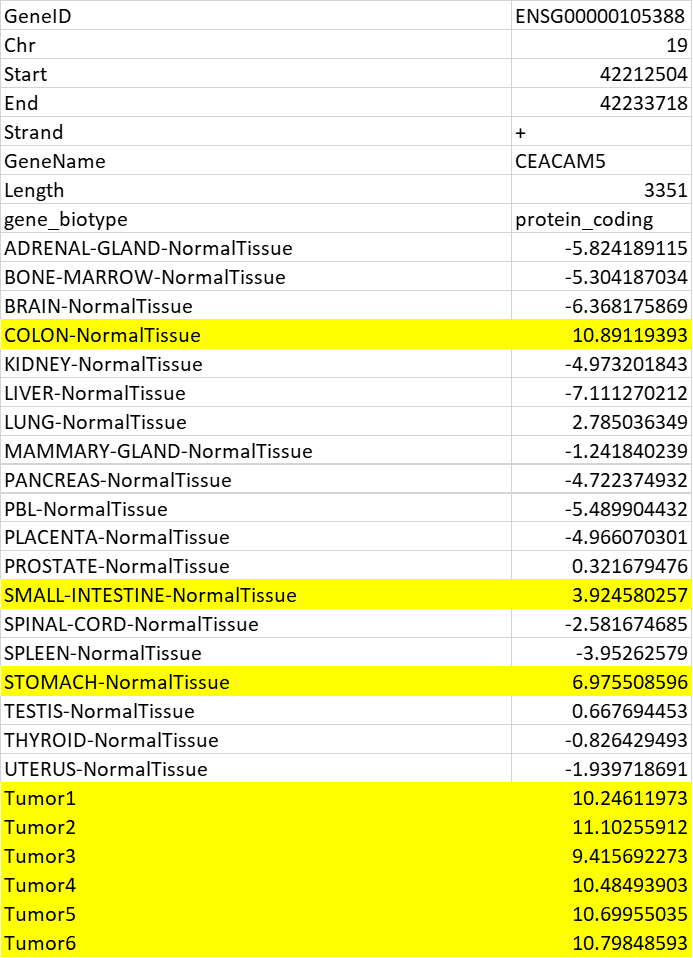
limma: Output
