clusterProfiler does exactly what you have done, and then passes those data on to the fgsea package, which then apparently filters out some of the genes. It's not clear to me when/why that happens, but maybe the maintainer of fgsea will provide an answer.
I should also point out that using Ensembl Gene IDs in this context is problematic. What will happen is the Ensembl Gene IDs will first be mapped to NCBI Gene IDs, and then those will be used to map to GO terms. This is unavoidable if you use an OrgDb, because the underlying database has NCBI Gene IDs as the central key, so any query between tables is mapped via the NCBI Gene ID. Anyway, the first step is quite frail, as EBI/EMBL and NCBI do not agree on which genes are the same, so you will silently drop genes at that first step.
As a super simple example, here's how many Ensembl Gene IDs cannot be mapped
> library(org.Mm.eg.db)
> mapper <- mapIds(org.Mm.eg.db, keys(org.Mm.eg.db), "ENSEMBL", "ENTREZID", multiVals = "list")
'select()' returned 1:many mapping between keys and columns
> nagns <- names(mapper)[sapply(mapper, function(x) all(is.na(x)))]
> length(nagns)
[1] 40079
> length(nagns)/length(mapper)
[1] 0.5484189
## Ooof
## how many of those NCBI Gene IDs does EBI/EMBL map?
> library(biomaRt)
> mart <- useEnsembl("ensembl","mmusculus_gene_ensembl")
> ensmap <- getBM(c("ensembl_gene_id","entrezgene_id"), "entrezgene_id", nagns, mart)
> head(ensmap)
ensembl_gene_id entrezgene_id
1 ENSMUSG00000102439 14246
2 ENSMUSG00000121135 15365
3 ENSMUSG00000070645 19702
4 ENSMUSG00000107355 100740
5 ENSMUSG00000120083 67644
6 ENSMUSG00000085385 68108
> dim(ensmap)
[1] 217 2
So NCBI only maps ~45% of their IDs to Ensembl IDs, and of those that don't map, Ensembl says 217 DO map. You can do the converse (use biomaRt to map Ensembl IDs to NCBI Gene IDs) and you will get a different set of genes that do/don't map.
Anyway, long story short, you would be better served if you could work with NCBI Gene IDs throughout.


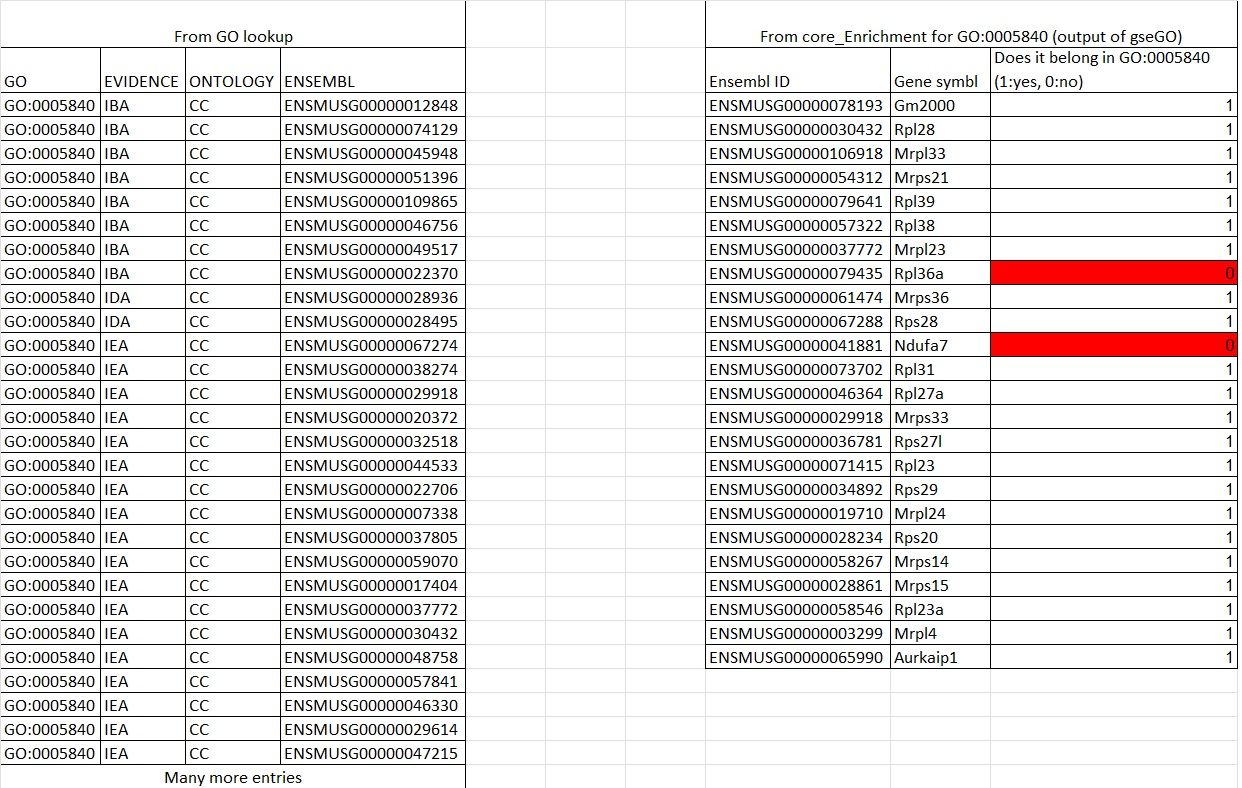
You indicated that you are able to find all genes associated with a GO term, but did you then filter out the genes that are not in your dataset?
Also, showing the code you used would be helpful.
I use:
I then get an csv that gives: Where I get the genes impacted in the set under core_enrichment, but this is only 15% of the genes in a set of 430. I want to find the other 365 terms that were not impacted. This would make a complete the list associated with the GO term specific to this gseGO function's GO term. But I have yet to find a way to give me that.
Where I get the genes impacted in the set under core_enrichment, but this is only 15% of the genes in a set of 430. I want to find the other 365 terms that were not impacted. This would make a complete the list associated with the GO term specific to this gseGO function's GO term. But I have yet to find a way to give me that.
If I use:
To get a complete list of mapped GO terms and ensembl IDs, the # of genes do not match the setSize when looking at a particular GO term and sometime not all the core_enrichment genes are listed.