Entering edit mode

Hi,
I have a raw count data set of a viral infection consisting of 3 time points and a virus free control with 3 replicates each produced with featureCounts.
Normalization after running template_script_DESeq2.r:
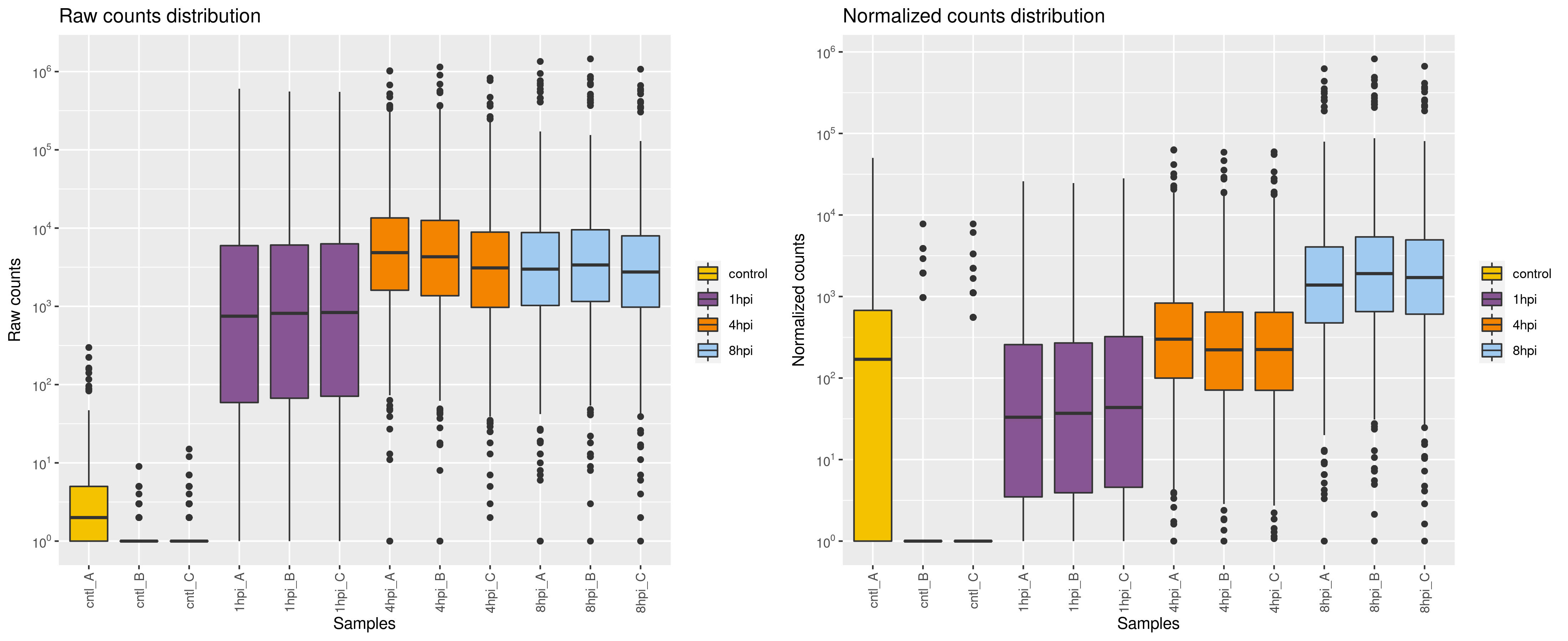
It does not look like the distributions across samples are stabilized. Apparently there were some problems with controls, especially replicate A. Since I am working only on the viral side of the replication cycle, I have just changed condRef in the template script from "control" to to one of the groups (a time point), thus dropping the controls. Normalization improved!
Is this the correct/intended way to do that?
Thank you!

Yes, these counts are viral genes alone. There is no host genome available yet...
I agree that you may have to drop the controls.
So isn't it correct for the controls to have almost no reads? Why would you want to do anything to make pretend that they have comparable counts to infected samples? The premise of size normalization; that some genes with median expression are unchanging; looks wrong for the viral genes alone, even in the non-control samples. Did your prep collect any host RNA? If so, I'm not sure it's right to proceed by totally ignoring that. Including it would probably make normalization work.
Yes, to have almost no viral reads in the virus free control is what one would expect. Since including the controls disturbs the normalization and the biological question (for now) is the expression dynamics of the viral side, it should be fine to drop them. The data I have produced this way makes more sense in the context of promoter and proteomics data...