Entering edit mode

Hi.
I am very new to bioinformatics/programming. Running the code for the first time ever using atena package (following their manual). I got the following error. What am I doing wrong? I have attached my full working screen's screen shot for reference.
Thanks a lot.
Lalani
''''r In addition: Warning message: In gzfile(file, "rb") : cannot open compressed file '', probable reason 'Invalid argument'
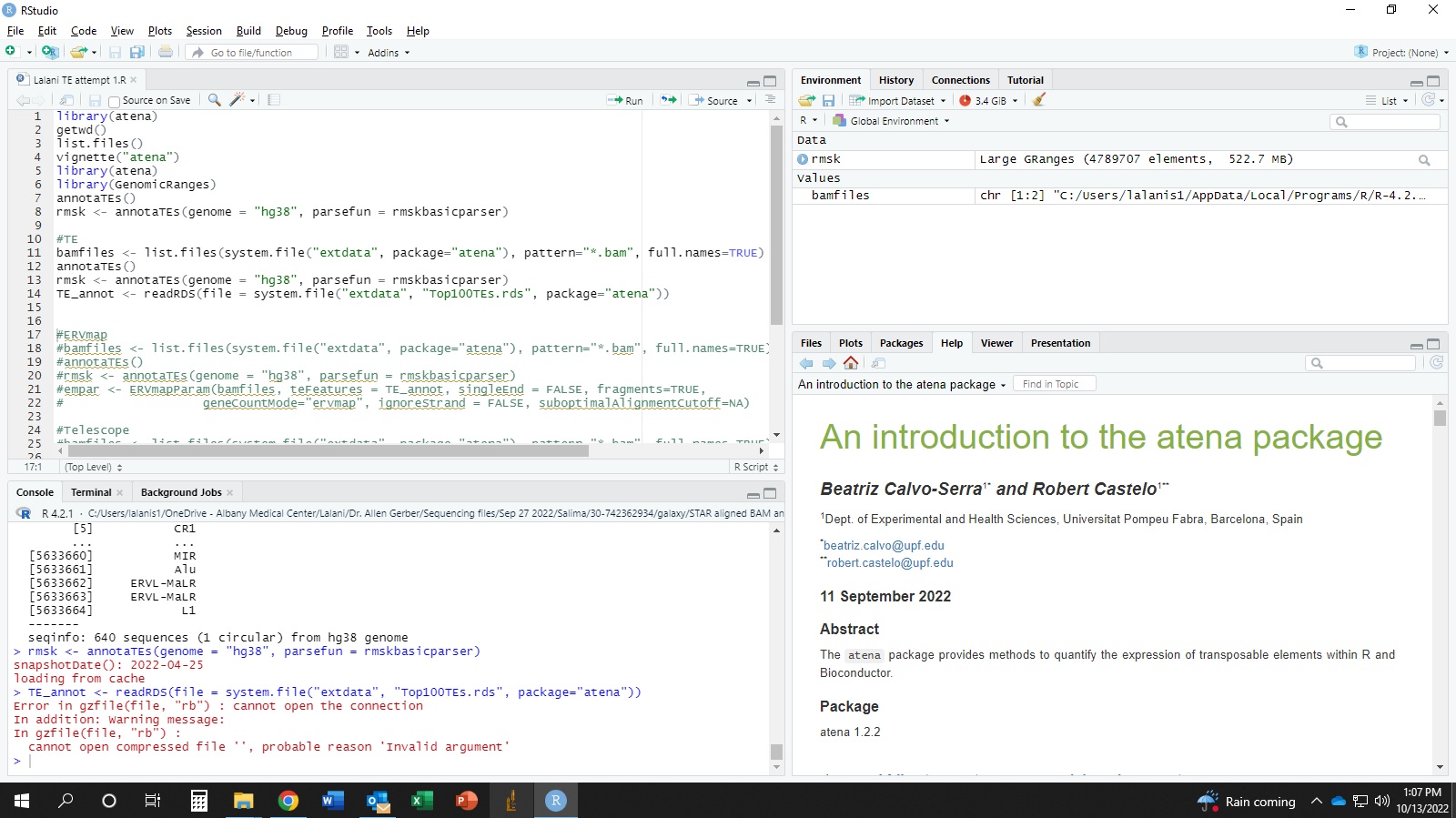
#problematic code
#TE
bamfiles <- list.files(system.file("extdata", package="atena"), pattern="*.bam", full.names=TRUE)
annotaTEs()
rmsk <- annotaTEs(genome = "hg38", parsefun = rmskbasicparser)
TE_annot <- readRDS(file = system.file("extdata", "Top100TEs.rds", package="atena"))
# results of running the following in an R session
> #TE
> bamfiles <- list.files(system.file("extdata", package="atena"), pattern="*.bam", full.names=TRUE)
> annotaTEs()
snapshotDate(): 2022-04-25
loading from cache
GRanges object with 5633664 ranges and 4 metadata columns:
seqnames ranges strand | swScore repName repClass
<Rle> <IRanges> <Rle> | <integer> <character> <character>
[1] chr1 10001-10468 + | 463 (TAACCC)n Simple_repeat
[2] chr1 15798-15849 + | 18 (TGCTCC)n Simple_repeat
[3] chr1 16713-16744 + | 18 (TGG)n Simple_repeat
[4] chr1 18907-19048 + | 239 L2a LINE
[5] chr1 19972-20405 + | 994 L3 LINE
... ... ... ... . ... ... ...
[5633660] chrX_KV766199v1_alt 179150-179234 - | 255 MIR1_Amn SINE
[5633661] chrX_KV766199v1_alt 184474-184785 - | 2039 AluJb SINE
[5633662] chrX_KV766199v1_alt 186964-187271 - | 386 MLT1G3 LTR
[5633663] chrX_KV766199v1_alt 187486-187569 - | 270 MLT1G3 LTR
[5633664] chrX_KV766199v1_alt 187597-187822 - | 1301 L1MA8 LINE
repFamily
<character>
[1] Simple_repeat
[2] Simple_repeat
[3] Simple_repeat
[4] L2
[5] CR1
... ...
[5633660] MIR
[5633661] Alu
[5633662] ERVL-MaLR
[5633663] ERVL-MaLR
[5633664] L1
-------
seqinfo: 640 sequences (1 circular) from hg38 genome
> rmsk <- annotaTEs(genome = "hg38", parsefun = rmskbasicparser)
snapshotDate(): 2022-04-25
loading from cache
> TE_annot <- readRDS(file = system.file("extdata", "Top100TEs.rds", package="atena"))
Error in gzfile(file, "rb") : cannot open the connection
In addition: Warning message:
In gzfile(file, "rb") :
cannot open compressed file '', probable reason 'Invalid argument'

Editted Hi.
Thank you so much for your help. I am working with human genome. I was able to get the hg38 from repeatmasker using
annotaTEs(genome = "hg38", parsefun = rmskidentity)
However, I am still getting the same error
I get it that my desired .rds is not present in atena package. Then what should be done to read top 100 TEs in human genome using atena?
Thanks.
Hi,
To quantify TE expression with the
atenapackage you do not need an object with annotations for the top "x" TEs (what would correspond to theTE_annotobject in the vignette). Maybe the vignette fromatenamay lead to confusion when we use annotations for only the top 28 expressed TEs for that specific dataset:we only use a subset of the annotations in the vignette to minimize the computational cost and memory requirements for building the vignette. These 28 TEs were selected after quantifying all TEs in RepeatMasker annotations and, then, selecting those top 28 expressed TEs.
To quantify TE expression with the
atenapackage, you need the TE annotations (you do not need the annotations of the top expressed TEs), which can be retrieved as you did using the command:Then, you have to build a parameter object (using the previously obtained TE annotations) and, finally, quantify TE expression with the
qtex()function. This will result in aRangedSummarizedExperimentobject with the expression levels of all TEs, which you can, then, use to identify the most expressed TEs in your samples (you can use it to identify the top 100 TEs in your samples, for example).In summary, you cannot use
atenato directly read the top 100 TEs in your dataset, you first need to quantify expression of all TEs and then identify the top expressed TEs.Thank you so much for your response. I will try to build parameter object (although I don't know what it is and how to do it at all but will work on it for sure). Hope I can do this.
I really appreciate your help and your patience with a person trying to use R and coding for the first time ever.
Best regards, Lalani
Hi.
I tried to build parameter object. I am having difficulty in doing so. Will it be possible for you to share an example of some other organism (if not human) so I can identify what parts of codes are required to be changed. This way, I will be able to relate it better and work to build parameter object for human genome.
Thanks.
This is what i was able to do using git hub code of yours. Not sure if it is ok at all. Please guide.
Hi Lalani,
Looking at the code from your last comment I think you are using as a guide the code found in
inst/scripts/create-data_OhtaniSaito13.Rmd, is that correct? If so, I am afraid that this code is not intended to serve as a guide or pipeline to carry out analyses with theatenapackage, instead, this filecreate-data_OhtaniSaito13.Rmdis only meant as a documentation on how the files used for the examples and the vignette (found ininst/extdata) were created.To use the
atenapackage to quantify TE expression, the code is much simple:First, you can retrieve TE annotations using the
annotaTEs()function available in theatenapackage. For instance if you want to quantify TE expression in.bamfiles from human aligned to hg38, you can:where the
genome=argument can be used to specify which genome version you want. For more information on this function you can check the manual.If instead you have already downloaded TE annotations and want to use a specific file you can read that file (for example using the
rtracklayerpackage) and create aGRangesorGRangesListobject.Second, you build the parameter object for one of the quantification methods available. For example, for the Telescope method:
Here, in
TelescopeParam()you need to define some arguments (singleEnd,ignoreStrand, etc.) depending on the type of.bamfiles you are using (if they are paired-end, stranded, etc.), you can read the manual to know more.Finally, you quantify TE expression using
qtex():This will output a
RangedSummarizedExperimentwith TE expression levels, you can access them by:and then you can use these expression levels to identify the top expressed TEs, or do differential expression analysis, etc.
I hope this answers your question!
Thanks a lot for explaining it in detail.
I have run it now. Hope it goes well. Will update here if anything goes wrong.
Thanks again.
Regards, Lalani
Hi.
We ran the package with some trials and errors. TE transcripts and ERVmap ran fine. However, we are getting the below error for telescope. We were wondering where should we make changes in the code to attend the recommended suggestions if you could kindly assist in this regard.
Many thanks.
Hi,
could you show us the result of calling:
and
right after you get the error?
thanks!
Hi
Sorry for the delay. Here are the requested results.
Thanks.
It seems the problem might be related to something in the BAM file that is not properly handled by the software. Would it be possible for you to identify the BAM file that causes the error, by running the software on just that BAM file and sharing that BAM file privately? (you can reach me by email at robert.castelo@upf.edu).
Sure. Will try to rule out and get back to you.
It is strange though as same bam files have worked for other two (ERVmap and TEtranscripts). Would that mean that the results for those two might not be accurate as well?
Not necessarily, in our own benchmarking (not yet published) the R implementation of the three methods reproduce very reliably their original implementation, so for us the question is whether the BAM file that causes this error has features that make it a cornercase outside our benchmarking, where we should correct something to address that problem, particularly in Telescope.
Thanks for your response. I have emailed you. I will need to check how I can share the bam file. Will get back to you on this.
Thanks again!
Hi Lalani,
We have fixed the bug that lead to the error. The new version of the package (with the bug fixed) will be available in 24-48h. To use it, you will need to re-install the package:
The error appeared due to the presence of not properly paired reads (e.g. with mates in different chromosomes). Thanks for bringing up this error, it has helped us to identify slight differences in the handling of this type of reads with respect to the algorithm used by the original Telescope method. We are still working on it to completely reproduce Telescope’s approach for not properly paired reads. However, this new version of the package provides a close implementation (in fact, since not properly paired reads make a very small fraction of the total number of reads, results should be very similar).
Thanks a lot, Calvo.
Will run my files with revised version soon!
Best regards, Lalani