Entering edit mode

Dear authors,
I came across your null ranges function. Kindly address my doubts.
I have a small RNA loci data and i want to test the association with other genomic feature. I want to generate bootstrapped data for my small RNA loci. One way of doing is to perform subsampling and another way is using bootRanges.
gr1 is my granges data representing my small RNA loci. I want to bootstrap only these regions because other regions are not small RNA loci. Therefore, i added the background genome coordinates to the exclude option (gr_toexclude). Is it right?
How do i select block length??? If you say i can share my loci data so that you can help me in deciding the block length.
p1 = bootRanges(gr1,
blockLength=100, R=100,
exclude=gr_to_exclude,
type="bootstrap",
withinChrom=TRUE)

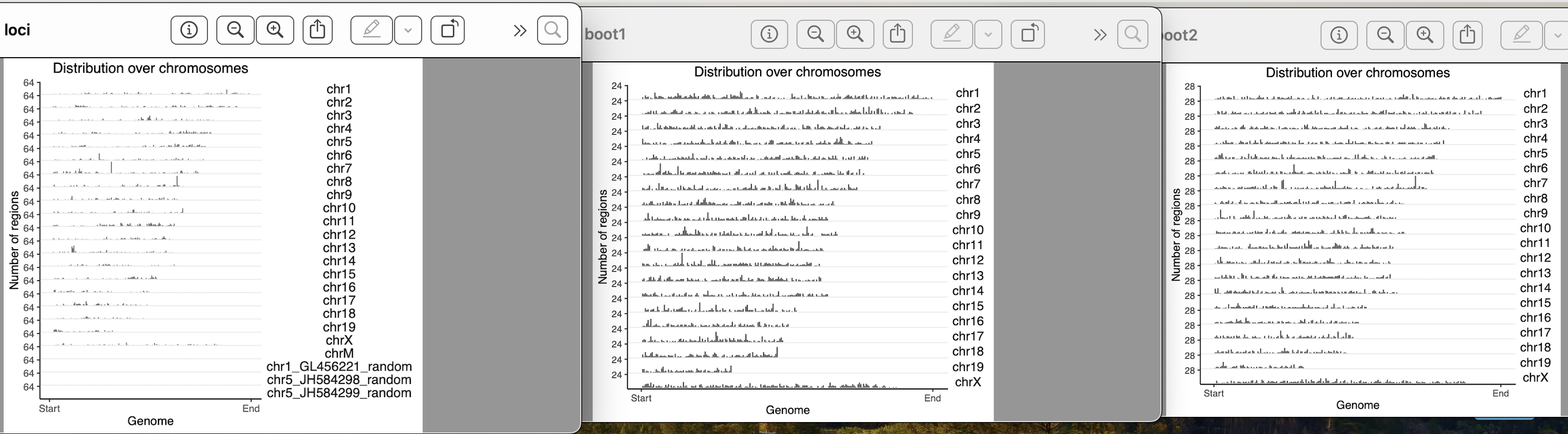
yes, they exist in clusters. How it should be done for a clustered data??
I used block length from 100bp, 5000bp, 10000bp, 100000bp, 1000000bps. As i am increasing the block length after 10000bp, the bootranges comes with zero width. Can you please explain me what this block length is doing?? what it should be for a clustered data??
Let's discuss a few of the points of bootRanges:
blockLength- you want this to be larger than the typical clustering pattern but smaller than the region you are tiling with blocks. We used a large segmentation of hg38, e.g. >1Mb contiguous segments, and found that 200-500 kb blocks worked well.seg- you can choose which blocks go where, or what parts of the genome to bootstrap. We typically bootstrap the entire genome, but you can bootstrap smaller segments only, I am still not sure where you want to place your bootstrapped features. Can you explain more about this choice for your data?exclude- you can additionally specify places that features should not be placed, in addition to how you specify the segmentation.Maybe you can give me an example of a locus and also the genomic context, and where you want the bootstrapped features to be placed. There is a lot of flexibility in this package, as there are many different use cases (e.g. whether the features live in genome space or transcript space, etc.).
Thank you for your response. I will share the data through email. I will share bootranges outcome also.
If you want a default analysis, you can use the example in the quick start:
https://nullranges.github.io/nullranges/articles/bootRanges.html#quick-start
Here we use a basic segmentation for hg38 and some recommended excluded regions, with block length of 500kb.
We bootstrap across chromosome typically (meaning a feature from chromosome A can be placed on chromosome B in the bootstrapping). Is there a reason why you don't want to do that?
I thought of keeping it the way it is, otherwise there is no specific reason for within chromsome bootstrapping.
I'd recommend to keep that argument as default:
withinChrom = FALSEOkay. I will do that.
Hello Dr. Michael,
I did the analysis with withinChrom=FALSE
How do you calculate inter range or inter feature distance? how do you take care of the different chromosomes? Because by any mean your boot ranges data does not look like your original data. In the original data the locus were apart and there were no clashes. In the bootranges generated data from one locus it has made three entries in different iters. If i take all iter to calculate inter range distance, it is less than the original. How should i calculate it?
We have a part of the bootRanges vignette where we assess the distance between subsequent ranges. Importantly, this is done per iteration, not across all iterations. It's probably sufficient to just look at one iteration, here we look at three:
https://nullranges.github.io/nullranges/articles/bootRanges.html#assessing-quality-of-bootstrap-samples
The bootstrapped ranges are supposed to look roughly similar in terms of this distribution to the original data, if everything has been set up correctly.
Thank you for your help Dr. Michael.
One more doubt, my original loci contains 7000 locus on different chromosomes. When i am generating bootranges one iteration has only 10-13 locus entries on different chromosomes. Is it okay?
The number should be roughly 7000 across the iterations.
Can you show table(seqnames(gr)) for the original and for one iteration of the bootstrap?
You can do
R=1if you want to just look at a single iteration.My guess is that you are losing >99% of the bootstrapped data to excluded regions. We thought of the excluded regions as e.g. telomere, centromere, just a small number of places where it doesn't make sense to put blocks.
What do you have for
sum(width(excluded))?It may help me to understand: how you are defining the excluded regions?
I excluded everything which was not locus. If small rna locus is from 2000 to 4000 and next locus is from 5000 to 6000, i excluded 1 to 2000 and then 4000 too 5000 and defined chr length as the last granges number for that chromosome.