Entering edit mode

Hi all,
I have been trying to extract the GSEA results from a list of genes after RNAseq analysis. It looks like my gseKEGG function is giving me problems. I am unable to generate a list of KEGG terms, it seems as my list of genes can't be mapped.
Could you help me please?
res_entrez <- dplyr::filter(res_ids, entrezid != "NA")
res_entrez <- res_entrez[which(duplicated(res_entrez$entrezid) == F), ]
foldchanges <- res_entrez$log2FoldChange
names(foldchanges) <- res_entrez$entrezid
foldchanges <- sort(foldchanges, decreasing = TRUE)
set.seed(123456)
gseaKEGG <- gseKEGG(geneList = foldchanges,
organism = "mmu",
nPerm = 1000,
minGSSize = 20,
pvalueCutoff = 0.05,
verbose = FALSE)
sessionInfo( )
This is the message I get when running the code:
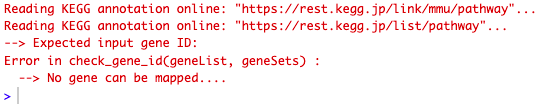
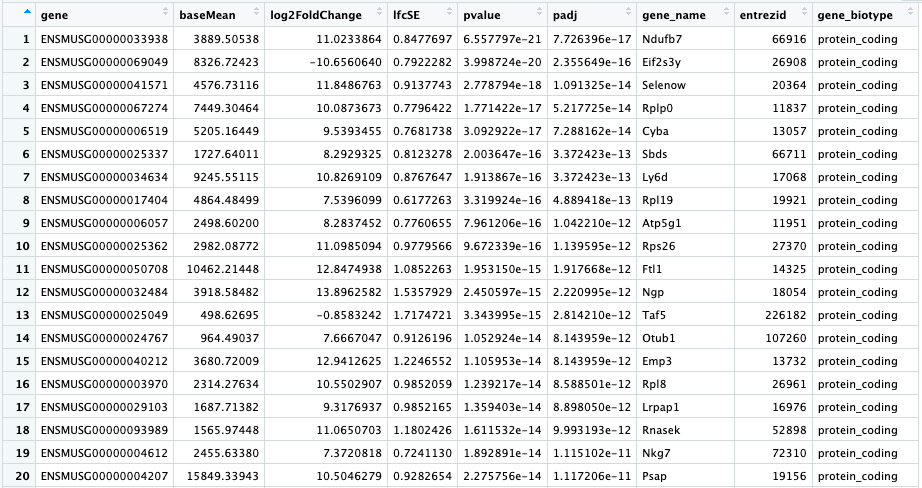
Here is what my "res_entrez" object looks like.
I also tried the test with DOSE package, it seems to work and the geneList seems similar.
data(geneList, package="DOSE")
kk2 <- gseKEGG(geneList = geneList,
organism = 'hsa',
minGSSize = 120,
pvalueCutoff = 0.05,
verbose = FALSE)
Here is what head of both geneList gives me


As input for
gseKEGGyou usedgeneList = foldchanges, so you should show the content of that numeric vector (and not ofres_entreznorfoldchanges_sex!).Also, as a side note, please use text, and do not paste pictures from R-studio... since that prevents easy copying of code or identifiers if needed.