
Is there any chance to solve "the model matrix is not full rank" in DESeq2?
My design matrix is a count matrix, one row per gene and one column per sample,
The sample information (colData in dds) is a dataframe with two column, a column for condition containing pre and post treatment and column containing subject Id.
I am asking this question with regards to do paired samples
The problem is happened when
dds_1 <- DESeqDataSetFromMatrix(countData = df, colData = sample_info, design = ~ subject + condition)
This is the error I get:
Error in checkFullRank(modelMatrix) : the model matrix is not full rank, so the model cannot be fit as specified. Levels or combinations of levels without any samples have resulted in column(s) of zeros in the model matrix.
I read the vignette section 'Model matrix not full rank': but I could not find any solution for the problem.
Is there any solution for this problem?


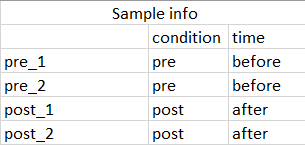
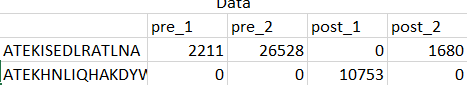
Thanks a billion for all of your kind help and time.