Entering edit mode

I'm working with a large data-set with multiple treatment time points and genotypes. To increase my sample size for one of the time points, I'd like to add in two samples that were collected and sequenced in a different run. Alignment methods are also different (STAR vs Illumina DRAGEN).
I’ve merged the counts table (I had a different number of total genes so I removed non-shared genes) and then ran RUVr to remove batch effects since it was recommended for my dataset regardless of adding in the new samples. The samples clustered nicely in the PCA (circled)only after running RUVr. Is this a suitable approach? Alternatively, would I have to correct for this in another way potentially using SVA or accounting for it in my design formula?
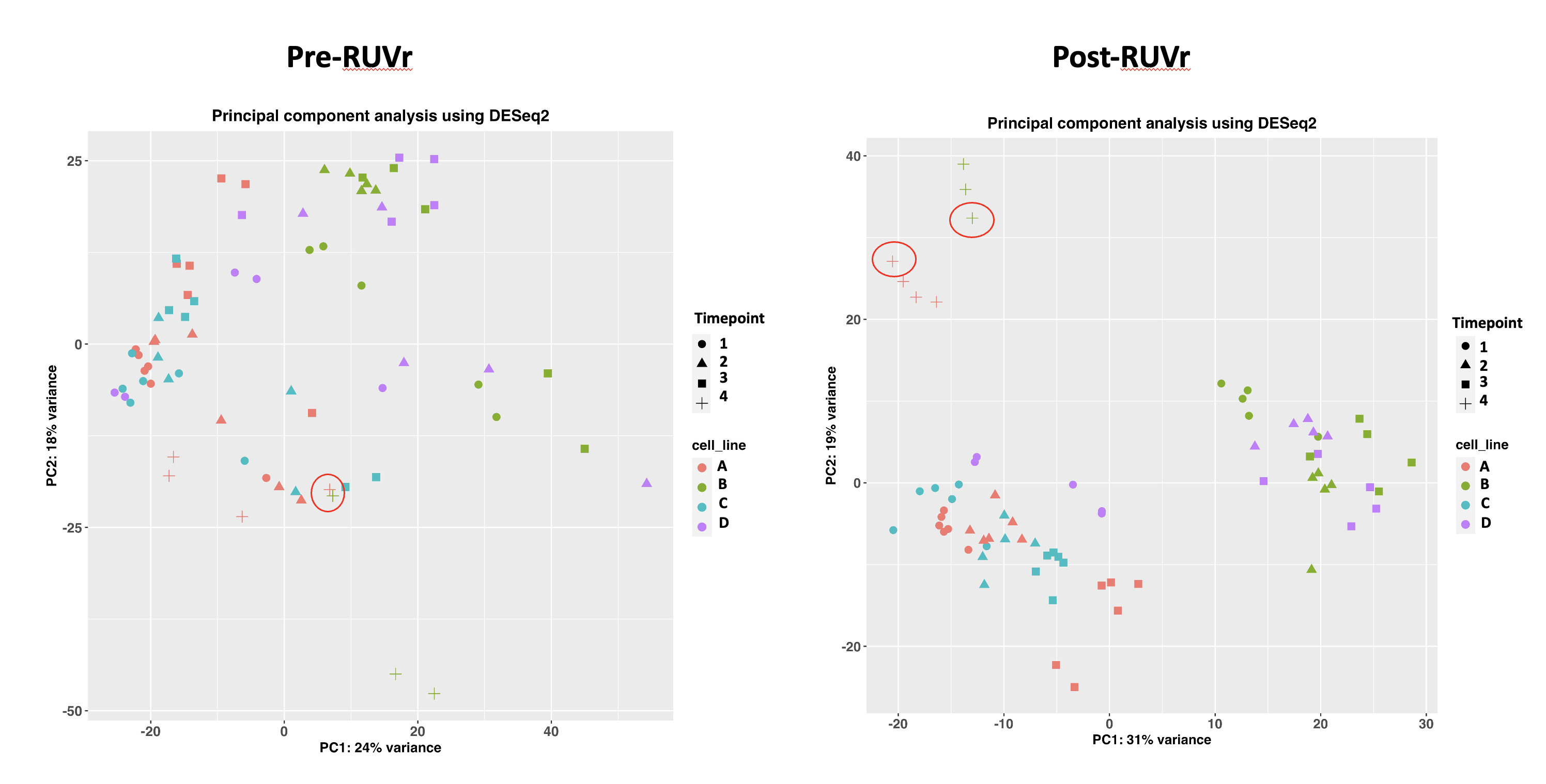

Thanks Michael! I've included the factors of unwanted variation to get the PCA plot on the right. Is that a suitable approach to combine replicates obtained from different sequencing runs?
Adding in these replicates also changes my DEG list. Is that because of a change in the model fit?
Sorry for delay, this got buried in a list of incoming messages.
Using the factors in the design formula is a good approach. It would be entirely expected that the DE list would change after controlling for technical variation.
Hi Michael, thank you so much for your responses that have guided me through my analysis. Sorry for the multiple follow-up questions, I have recently tried to implement Combat-seq to combine the different sequencing runs. Here, I first use combat-seq to combine the runs accounting for the known batch effect, followed by RUVr to remove unknown batch effects as opposed to above where I run RUVr alone.
Would I be over-correcting by using Combat-seq followed by RUVr (the DEG list is really affected). Should I stick with only using RUVr to combine sequencing runs and remove unknown batch effects? I'm unsure of the best option to proceed with, would you have any advice?