Entering edit mode

Hi all,
Does positive fold in this case mean the gene in the diseased group is more accessible than the control group? The object is created from the DiffBind package. Thank you so much!
Hi all,
Does positive fold in this case mean the gene in the diseased group is more accessible than the control group? The object is created from the DiffBind package. Thank you so much!

Yes, as you've set up the experiment, positive fold changes indicate more accessible chromatin in the diseased state (higher read concentration, shown in log2 of normalized overlapping read counts) than in the control state (lower read concentration). Hopefully this is reflected in what you can see in a genome browser at these locations.
Use of this site constitutes acceptance of our User Agreement and Privacy Policy.
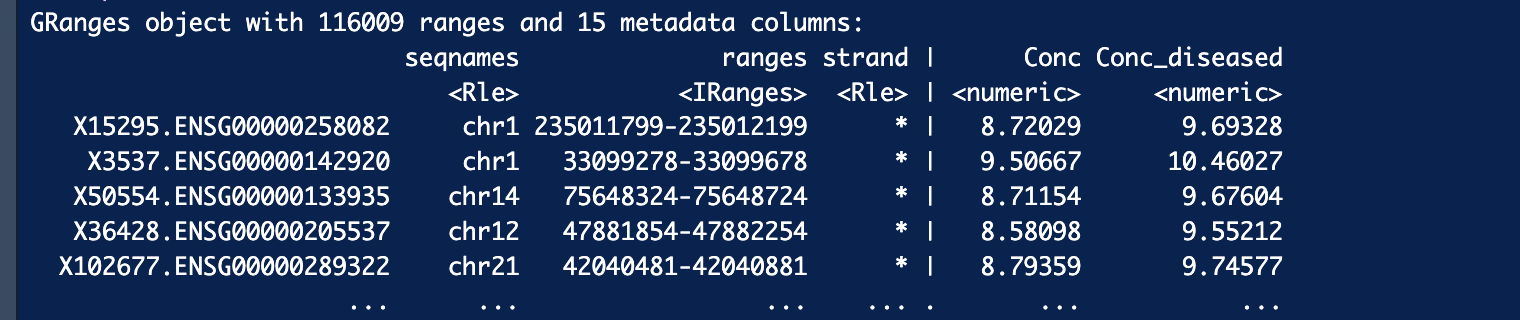

Thank you so much for your reply! I converted Ensemble ID to gene ID and after filtering out NA and keeping unique genes, I still have about 60k genes. Is that unusual?
It is not that unusual to see tens of thousands of differences in open chromatin regions if the two sample groups are quite different tissues. However, considering there are fewer than 25k genes in the human genome this seems like a lot! It might be more interesting to see what genes aren't in your list.
Thank you so much for the suggestion! Two sample groups are the same cell types. There are should be more genes closed than the number of genes accessible, is that right? Or maybe something was wrong in my analysis which made the number of genes too high. My correction, after keeping only unique genes, I have about 21k genes which are still too high. Would you suggest possible reasons for that? Your manual has only 246 ranges.
The sample dataset used in the vignette uses only one (smaller) chromosome, and is a transcription factor (not ATAC), which is why there are only a few hundred DB sites. with ATAC-seq, having 20k (or more) differential sites genome-wide is not unusual.
I'm not sure exactly how you're mapping sites to genes, I was just surprised at how many genes were identified given that there are <25k genes total in the human genome.
I used ChIPpeakAnno::addGeneIDs to get the genes. Nf-core ATAC-seq pipeline to get bam files and broadpeak files.