Entering edit mode

Hello all,
From my understanding of the featureCounts manual it should, by default, count reads that align to the features (exons) of a meta-feature (gene). However my output file is at the exon level (sorry for the line formatting in the screenshot):

I can't tell if this is an issue with featurecounts, or my understanding of how my command should be formatted, or perhaps an issue with the feature IDs in my GTF file.
Here is the code I ran:
featureCounts -T 8 -t exon -g ID -a AmbTr_v01.0.gff.gff3 -o iwgc_counts.txt *.sam
and here is a snippet of my GTF file:
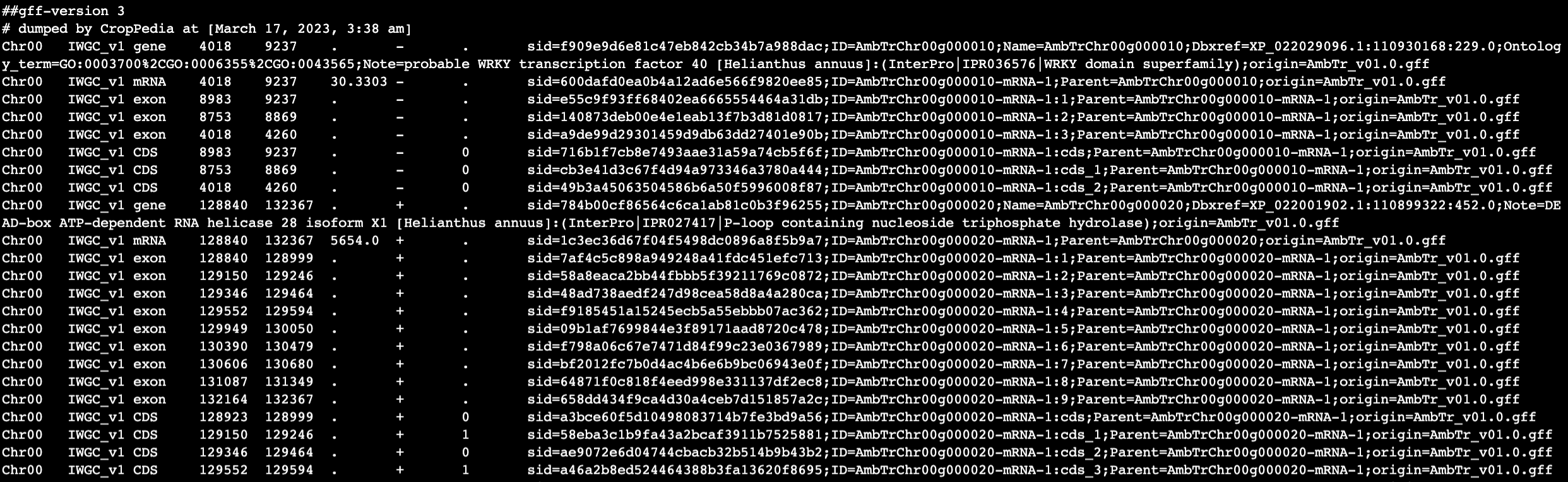
Any advice, explanation of what I am missing, or guidance would be greatly appreciated!
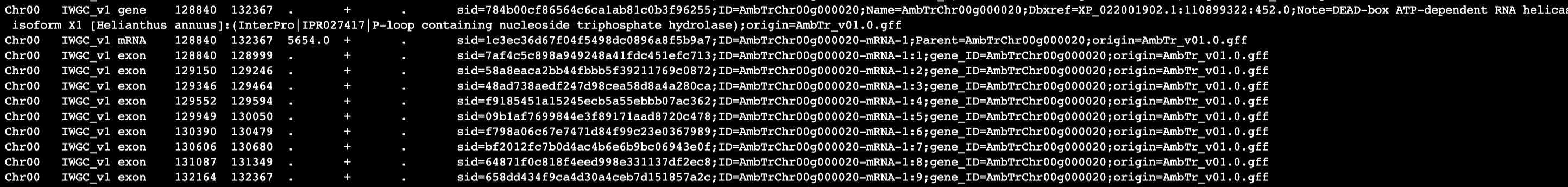

I don't know if this information will help either troubleshoot the issue or make it more obvious if I am misunderstanding the options, but here is a small update.
I used sed to change "ID" in gene rows to "gene_ID" to distinguish from IDs of other features, so now it looks like:
I then re-ran featureCounts with this command:
And from that I get this error: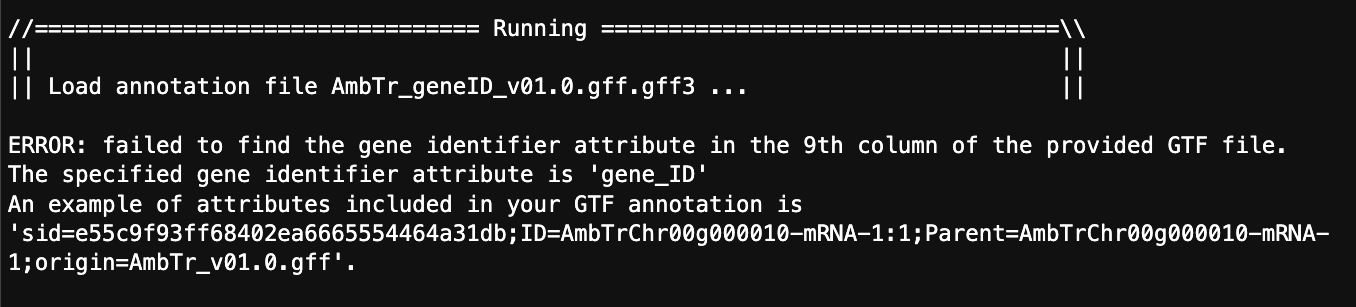
So, with these options should featureCounts be looking for the exon ID at this step or the gene ID?
Am I just massively misunderstanding the command?
I do not think I want to use -t gene because then it will count reads that aligned to introns as well, correct?
How can I get the total counts per gene, but only based on the reads aligned exons?
Please help!