
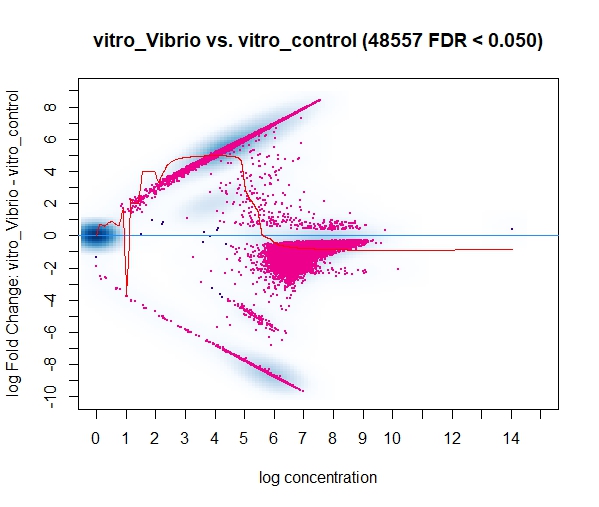
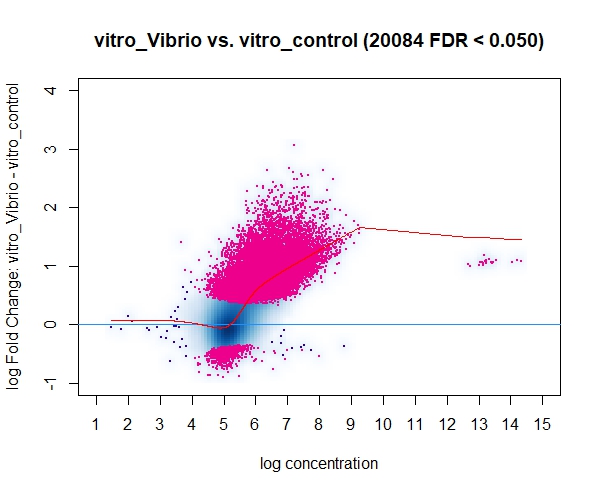
Currently I'm running DiffBind using bam/bed files that have been filtered to keep only the highly reproducible peaks (HR-peaks), obtained using the IDR method (for reference: https://hbctraining.github.io/Intro-to-ChIPseq/lessons/07_handling-replicates-idr.html), obtaining a good amount of DB peaks.
However, in a discussion with my colleagues a point was raised mentioning that the input for DiffBind should be all peaks on the dataset and not only the HR ones, resulting in way less DB peaks for some of our marks/conditions when using all the peaks (and not only the HR ones). I find this a little bit conflicting with what I know of DeSeq2 (which, if I understood it correctly, Diffbind uses some routines carried out by DeSeq2 too), so I was wondering if you could clarify this question? I assumed that I could use the HR peaks from the line below (found in the DiffBind vignette, page 4): (..) Peaksets are derived either from ChIP-Seq peak callers, such as MACS, or using some other criterion (...)
I'm running DiffBind with mostly default parameters but just in case here is the code:
peaksets <- dba(sampleSheet = samples)
readcount <- dba.count(peaksets)
normalized <- dba.normalize(readcount)
comparisson <- dba.contrast(normalized, contrast = c("Factor","challenge","control"), minMembers = 2)
diffanalysis <- dba.analyze(comparisson, bParallel = FALSE, bBlacklist = FALSE, bGreylist = FALSE)
Thank you so much for your time!
Óscar

This seems at odds with what I understand about DeSeq2 (and, if I'm not mistaken, Diffbind also makes use of certain DeSeq2–carried–out algorithms), so I was hoping you might shed some light on the matter. I had first thought that the HR peaks from the following line (page 4 of the DiffBind vignette) may be used: (..) In ChIP-Seq, peak callers like MACS are used to identify peaks, and these are then used as one of several criteria to generate peaksets. drift hunters
Your essay was beneficial to me, and I found it to be very useful. Thank you for writing it. You give a wealth of knowledge that is not only impressive but also insightful in the writing that you submit. You have my gratitude for having written that down. cookie clicker
This is a great question using all peaks vs. only HR peaks in DiffBind can definitely impact your differential analysis. Understanding how it interfaces with DESeq2 is key. While refining your workflows on Android, explore ApkTuti.com for free Game & App Apk download and trusted APK Downloads
Below is a detailed explanation of the considerations for using filtered versus Sprunked unfiltered input data in DiffBind, addressing the concerns raised in your query.
I'm glad to see that you have a unique way of writing the post. Now it's easy for me to understand the idea and put it into practice io games