Entering edit mode

I'm using DiffBind for ATAC-seq DA analysis. The MA plot and volcano look nothing like I expect. I do not expect a large difference between my experimental (FRDA) and control group. But as you can see in the figures, below, there is a heavy skew for regions down regulated compared to control.
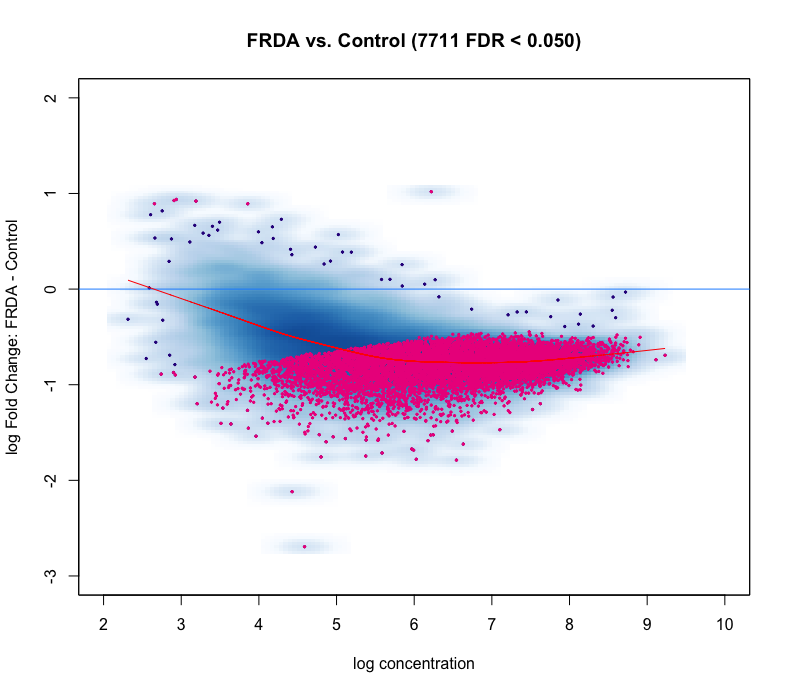
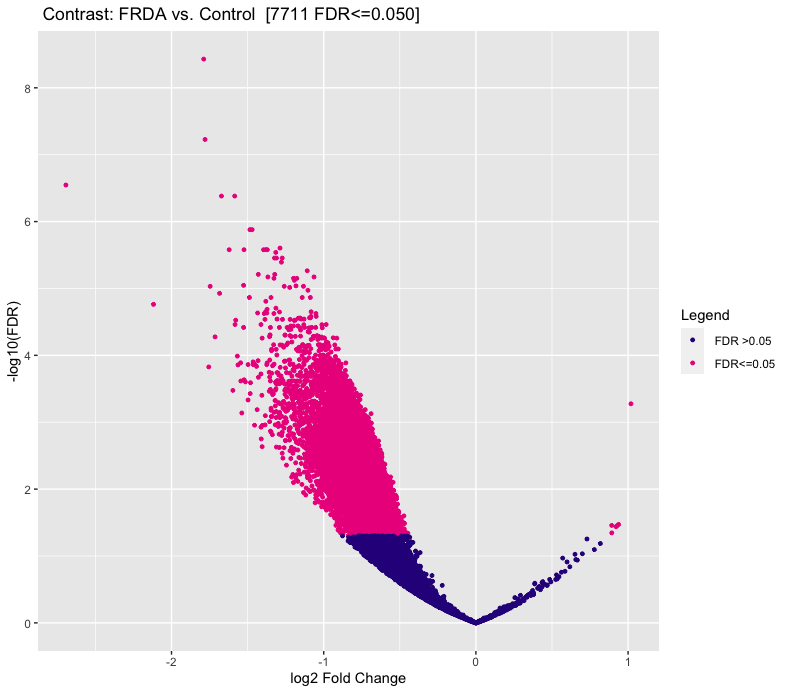
I tried loess curve offset normalization, since the curve doesn't look centered at all
counts_NP.loess <- dba.normalize(counts_NP, method = DBA_ALL_METHODS ,offsets = TRUE).
After adjusting the offset, the plots look even worse!
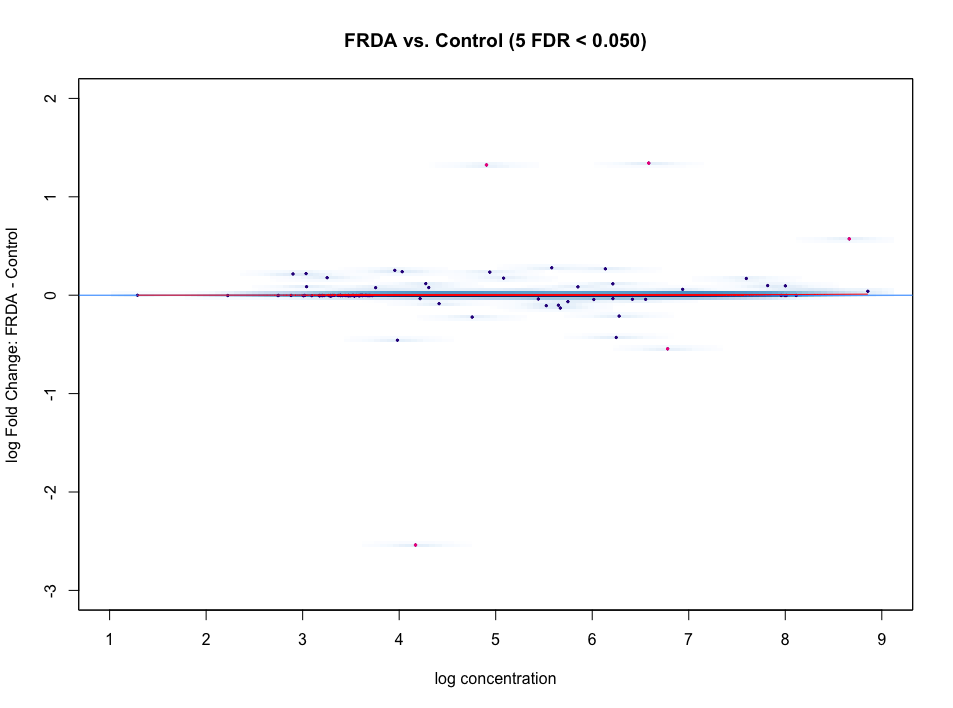
I'm fairly new to both ATAC and DiffBind, so any help and tips would be greatly appreciated!
> sessionInfo()
R version 4.2.1 (2022-06-23)
Platform: x86_64-apple-darwin17.0 (64-bit)
Running under: macOS Ventura 13.4
Matrix products: default
LAPACK: /Library/Frameworks/R.framework/Versions/4.2/Resources/lib/libRlapack.dylib
locale:
[1] en_US.UTF-8/en_US.UTF-8/en_US.UTF-8/C/en_US.UTF-8/en_US.UTF-8
attached base packages:
[1] stats4 stats graphics grDevices utils datasets methods base
other attached packages:
[1] csaw_1.30.1 GreyListChIP_1.28.1 DiffBind_3.6.5 SummarizedExperiment_1.28.0
[5] Biobase_2.58.0 MatrixGenerics_1.10.0 matrixStats_0.63.0 GenomicRanges_1.50.1
[9] GenomeInfoDb_1.34.4 IRanges_2.32.0 S4Vectors_0.36.1 BiocGenerics_0.44.0
loaded via a namespace (and not attached):
[1] utf8_1.2.3 spatstat.explore_3.2-1 reticulate_1.28 tidyselect_1.2.0
[5] RSQLite_2.3.1 AnnotationDbi_1.60.0 htmlwidgets_1.6.2 grid_4.2.1
[9] BiocParallel_1.32.4 Rtsne_0.16 munsell_0.5.0 codetools_0.2-19
[13] ica_1.0-3 interp_1.1-4 systemPipeR_2.2.2 future_1.32.0
[17] miniUI_0.1.1.1 withr_2.5.0 spatstat.random_3.1-5 colorspace_2.1-0
[21] progressr_0.13.0 knitr_1.43 rstudioapi_0.14 Seurat_4.3.0
[25] ROCR_1.0-11 tensor_1.5 listenv_0.9.0 labeling_0.4.2
[29] bbmle_1.0.25 GenomeInfoDbData_1.2.9 mixsqp_0.3-48 hwriter_1.3.2.1
[33] polyclip_1.10-4 farver_2.1.1 bit64_4.0.5 coda_0.19-4
[37] parallelly_1.36.0 vctrs_0.6.2 generics_0.1.3 xfun_0.39
[41] R6_2.5.1 apeglm_1.18.0 invgamma_1.1 locfit_1.5-9.7
[45] cachem_1.0.8 bitops_1.0-7 spatstat.utils_3.0-3 DelayedArray_0.24.0
[49] promises_1.2.0.1 BiocIO_1.8.0 scales_1.2.1 gtable_0.3.3
[53] globals_0.16.2 goftest_1.2-3 rlang_1.1.1 splines_4.2.1
[57] rtracklayer_1.58.0 lazyeval_0.2.2 spatstat.geom_3.2-1 yaml_2.3.7
[61] reshape2_1.4.4 abind_1.4-5 httpuv_1.6.11 tools_4.2.1
[65] ggplot2_3.4.2 ellipsis_0.3.2 gplots_3.1.3 RColorBrewer_1.1-3
[69] ggridges_0.5.4 Rcpp_1.0.10 plyr_1.8.8 zlibbioc_1.44.0
[73] purrr_1.0.1 RCurl_1.98-1.12 deldir_1.0-9 pbapply_1.7-0
[77] ashr_2.2-54 cowplot_1.1.1 zoo_1.8-12 SeuratObject_4.1.3
[81] ggrepel_0.9.3 cluster_2.1.4 magrittr_2.0.3 data.table_1.14.8
[85] scattermore_1.1 lmtest_0.9-40 RANN_2.6.1 truncnorm_1.0-9
[89] mvtnorm_1.1-3 SQUAREM_2021.1 amap_0.8-19 fitdistrplus_1.1-11
[93] patchwork_1.1.2 mime_0.12 evaluate_0.21 xtable_1.8-4
[97] XML_3.99-0.14 emdbook_1.3.12 jpeg_0.1-10 gridExtra_2.3
[101] compiler_4.2.1 bdsmatrix_1.3-6 tibble_3.2.1 KernSmooth_2.23-21
[105] crayon_1.5.2 htmltools_0.5.5 later_1.3.1 geneplotter_1.76.0
[109] tidyr_1.3.0 DBI_1.1.3 MASS_7.3-60 ShortRead_1.54.0
[113] Matrix_1.5-4.1 cli_3.6.1 parallel_4.2.1 metapod_1.4.0
[117] igraph_1.4.3 pkgconfig_2.0.3 GenomicAlignments_1.34.0 numDeriv_2016.8-1.1
[121] sp_1.6-0 plotly_4.10.1 spatstat.sparse_3.0-1 annotate_1.76.0
[125] XVector_0.38.0 stringr_1.5.0 digest_0.6.31 sctransform_0.3.5
[129] RcppAnnoy_0.0.20 spatstat.data_3.0-1 Biostrings_2.66.0 rmarkdown_2.21
[133] leiden_0.4.3 uwot_0.1.14 edgeR_3.38.4 restfulr_0.0.15
[137] shiny_1.7.4 Rsamtools_2.14.0 gtools_3.9.4 rjson_0.2.21
[141] lifecycle_1.0.3 nlme_3.1-162 jsonlite_1.8.4 viridisLite_0.4.2
[145] limma_3.52.4 BSgenome_1.66.1 fansi_1.0.4 pillar_1.9.0
[149] lattice_0.21-8 KEGGREST_1.38.0 fastmap_1.1.1 httr_1.4.6
[153] survival_3.5-5 glue_1.6.2 png_0.1-8 bit_4.0.5
[157] stringi_1.7.12 blob_1.2.4 DESeq2_1.38.1 memoise_2.0.1
[161] latticeExtra_0.6-30 caTools_1.18.2 dplyr_1.1.2 irlba_2.3.5.1
[165] future.apply_1.11.0
