
Hi Bioconductor folks,
this is my first question here, so let me know if you need any more specifics.
I have a bulk RNAseq data set of patient and healthy donor samples from different skin diseases. -Patients with 3 different diseases were included: AD, PSO or chronic AD; as well as a control healthy cohort. -Each patient gave two biopsies, lesional and non-lesional skin. -This means that for all patients there are 2 samples and for each healthy control there is 1
My metadata looks like this:
condition -> combining disease and patient id (e.g. AD_lesional, AD_non_lesional, PSO_lesional, ..., healthy)
patient.ID -> a number counting up, but resetting in each disease (for AD_004 lesional -> 1, for AD_004 non_lesional -> 1, for AD_005 lesional -> 2,..., for PSO_001 lesional -> 1, ...)
disease -> staing the disease (i.e. AD, PSO, chronicAD or Healthy)
sample.type -> staiting the state of the sample if its lesional or non_lesional
patient.id -> a number counting up but not resetting in each disease (for AD_004 lesional -> 101, for AD_004 non_lesional -> 101, for AD_005 lesional -> 102,..., for PSO_001 lesional -> 201, ...)
In total there are 21 AD patients (42 samples), 38 healthy controls (38 samples), 28 PSO patients (55 samples, one only gave 1 samples), and 6 chronicAD patients (12 samples).
My question would be the design formula to use for DESeq2. I thought i would need something like "design = ~ condition + condition:sample.type + patient.id" Thereby investigating the expression changes depending on the condition, but accounting for variations that arise from the sample type (inflamed/ lesional skin has a very different biology from non-inflamed/ non-lesional skin) and accounting for batch variations by using the patient.id.
The Error is, that the model is not full rank.
I could use a workaround and simply use "design = ~ condition" and still see the PCA cluster fine:
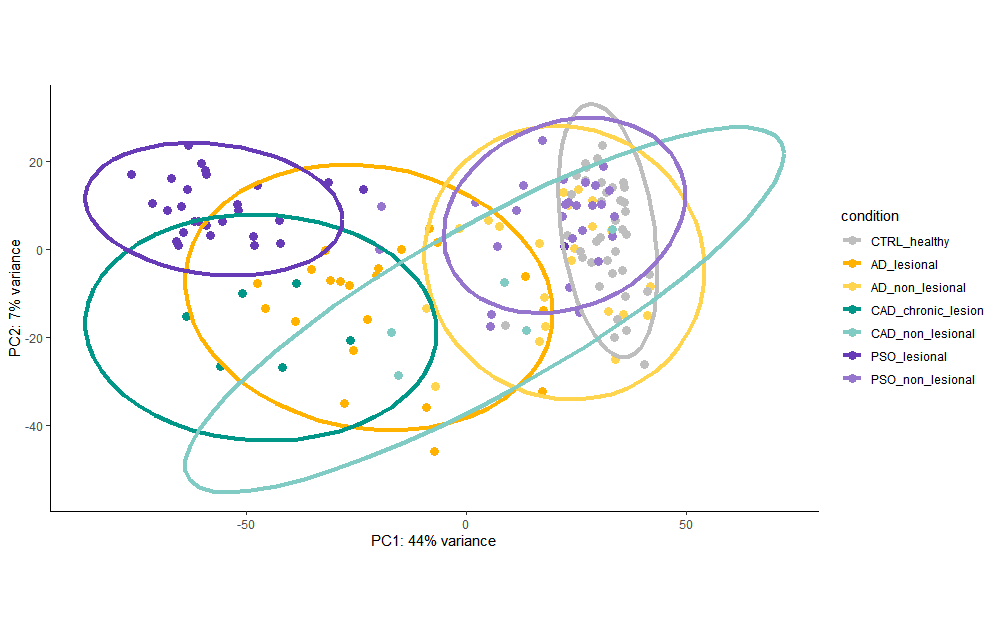
But i'm worried that this is not correct. I didnt get the workaround solution from the vignette to remove zeroes to work:
View(model.matrix(~ condition, meta_data)) #-> looks good
View(model.matrix(~ condition + condition:sample.type + patient.id, meta_data))
dds$condition
dds$condition <- droplevels(dds$condition)
dds$condition
m1 <- model.matrix(~ condition + condition:sample.type + patient.id, meta_data)
colnames(m1)
unname(m1) #just shows the matrix w/o colnames
all.zero <- apply(m1, 2, function(x) all(x==0)) #into variable all.zero, apply
# a function to check everything for values of 0, into the columns("2") of m1
idx <- which(all.zero) # store info: "which values in df are TRUE" into var idx
m1 <- m1[,-idx]
View(unname(m1))
dim(m1) #dim of m1 shrink from 147 x 376 to 147 x 103
dds_res_full <- DESeq(dds, full = m1)
Error:
> dds_res_full <- DESeq(dds, full = m1)
using supplied model matrix
Error in checkFullRank(full) :
the model matrix is not full rank, so the model cannot be fit as specified.
One or more variables or interaction terms in the design formula are linear
combinations of the others and must be removed.
Honestly i'm a little lost and would be super glad if you could help me. I kinda see the healthy control group as the problem because they only have one sample each. Meaning any info is kind of redundant. The model works with "design = ~ condition + patient.ID" but this leaves the patient number 1 assigned to the first samples of each group, which is not right. Am i right?
Thanks a lot in advance!

Hello James. Thank you for your reply. I guess you are referring to the section "Group-specific condition effects, individuals nested within groups"? It should apply to my case, but what I'm concerned about, it that in the solution, there are multiple "individual 1". One per group, as in my variable "patient.ID". Does DESeq2 now assume, that each ind. 1 is the same and would assume -so to say- paired values and does this impact the analysis?
Also applying the vignette solution, I would use the design formula " design = ~ patient.ID + condition + condition:sample.type". But with this I will always run into the problem that the model is not full rank, because CTRL_healthy has only one sample type and are therefore linear combinations of each other. Is there any way to circumvent this?
For your first question, no, DESeq2 will not assume all those Ind 1 are the same individual. You can see that's true for yourself by looking at the resulting design matrix (part of that section).
Note that there is a column for grpX:ind.n2, and a separate column for grpY:ind.n2. Those are the two individuals with Ind == 2 that are in separate groups.
As for your second question, the controls in this context are only useful for a non-paired analysis. In other words, any comparison to controls will be unpaired because the controls are different subjects, and you can make any comparison between your AD, CAD, or PSO subjects (either leisonal or not) to controls. But that's a separate comparison from your paired comparisons and will have to be done separately.