Entering edit mode

Hello, I am running differential expression analysis on age-related changes in transcription using natural splines with DESeq2 like so:
dds <- DESeqDataSetFromMatrix(countData = counts,
colData = coldata,
design = ~ ns(age_scaled, df = 3))
keep <- rowSums(counts(dds) >= 10) >= 3
dds <- dds[keep,]
dds <- DESeq(dds, test="LRT", reduced = ~ 1)
res <- results(dds)
And later plotting the fitted splines like so:
# Matrices for fitted curves:
coef_mat <- coef(dds)
design_mat <- model.matrix(design(dds), colData(dds))
dat <- plotCounts(dds, gene, intgroup = c("age", "sex", "genotype"), returnData = TRUE) %>%
mutate(logmu = design_mat %*% coef_mat[gene,],
logcount = log2(count + 1))
ggplot(dat, aes(age, logcount)) +
geom_point(aes(color = age, shape = genotype), size = 2) +
geom_line(aes(age, logmu), col="#FF7F00", linewidth = 1.2) +
labs(
title = gene,
x = "Age",
y = "Log2 expression count",
color = "Age"
)
I clustered the genes with kmeans and now apart from profiling the clusters I would like to visualize them, similar to an approach here:
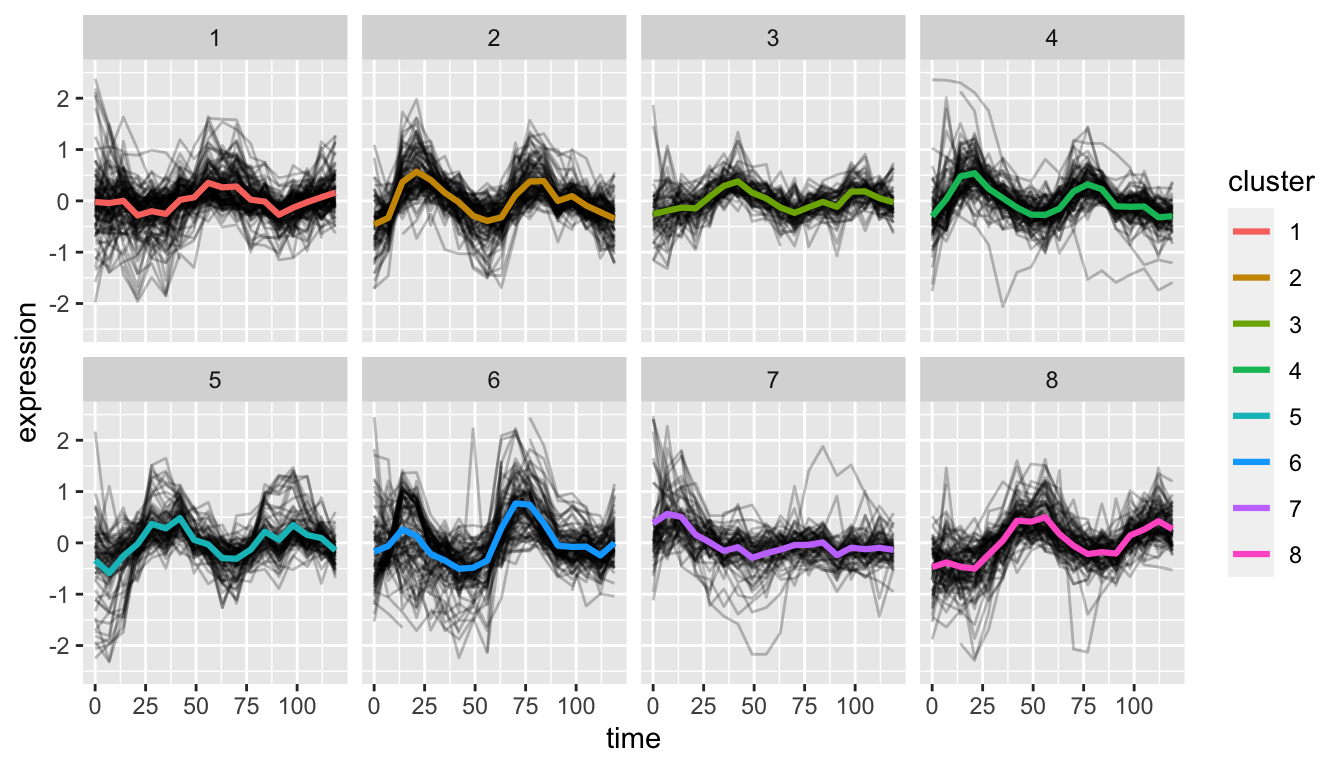 from: https://bio723-class.github.io/Bio723-book/clustering-in-r.html#depicting-the-data-within-clusters
from: https://bio723-class.github.io/Bio723-book/clustering-in-r.html#depicting-the-data-within-clusters
But in my case I do not have per group LFC as I was doing LRT. Any advice/ideas if I could still do a similar vizualization; extracting the splines per gene in my gene_list and perhaps putting Z-scores on the y-axis? I am struggling to figure out a way to reapply the code to my data. Thanks a lot in advance.
sessionInfo( )
R version 4.3.1 (2023-06-16 ucrt)
Platform: x86_64-w64-mingw32/x64 (64-bit)
Running under: Windows 11 x64 (build 22621)
Matrix products: default
locale:
[1] LC_COLLATE=English_United States.utf8 LC_CTYPE=English_United States.utf8
[3] LC_MONETARY=English_United States.utf8 LC_NUMERIC=C
[5] LC_TIME=English_United States.utf8
time zone: Europe/Berlin
tzcode source: internal
attached base packages:
[1] splines stats4 stats graphics grDevices utils datasets methods base
other attached packages:
[1] gplots_3.1.3 cluster_2.1.4 dendextend_1.17.1
[4] umap_0.2.10.0 vsn_3.68.0 shiny_1.7.5
[7] xlsx_0.6.5 readxl_1.4.3 EnhancedVolcano_1.18.0
[10] ggrepel_0.9.3 RColorBrewer_1.1-3 pheatmap_1.0.12
[13] DESeq2_1.40.2 SummarizedExperiment_1.30.2 Biobase_2.60.0
[16] MatrixGenerics_1.12.3 matrixStats_1.0.0 GenomicRanges_1.52.0
[19] GenomeInfoDb_1.36.1 IRanges_2.34.1 S4Vectors_0.38.1
[22] BiocGenerics_0.46.0 lubridate_1.9.2 forcats_1.0.0
[25] stringr_1.5.0 dplyr_1.1.2 purrr_1.0.2
[28] readr_2.1.4 tidyr_1.3.0 tibble_3.2.1
[31] ggplot2_3.4.3 tidyverse_2.0.0 BiocManager_1.30.22
loaded via a namespace (and not attached):
[1] bitops_1.0-7 gridExtra_2.3 rlang_1.1.1 magrittr_2.0.3
[5] compiler_4.3.1 png_0.1-8 systemfonts_1.0.5 vctrs_0.6.3
[9] pkgconfig_2.0.3 crayon_1.5.2 fastmap_1.1.1 XVector_0.40.0
[13] ellipsis_0.3.2 labeling_0.4.3 caTools_1.18.2 utf8_1.2.3
[17] promises_1.2.1 rmarkdown_2.25 tzdb_0.4.0 preprocessCore_1.62.1
[21] ragg_1.2.6 bit_4.0.5 xfun_0.40 zlibbioc_1.46.0
[25] cachem_1.0.8 jsonlite_1.8.7 later_1.3.1 DelayedArray_0.26.7
[29] xlsxjars_0.6.1 BiocParallel_1.34.2 parallel_4.3.1 R6_2.5.1
[33] bslib_0.5.1 stringi_1.7.12 reticulate_1.34.0 limma_3.56.2
[37] jquerylib_0.1.4 cellranger_1.1.0 Rcpp_1.0.11 knitr_1.44
[41] httpuv_1.6.11 Matrix_1.5-4.1 timechange_0.2.0 tidyselect_1.2.0
[45] viridis_0.6.4 rstudioapi_0.15.0 abind_1.4-5 yaml_2.3.7
[49] codetools_0.2-19 affy_1.78.2 curl_5.0.2 lattice_0.21-8
[53] withr_2.5.1 askpass_1.2.0 evaluate_0.22 rJava_1.0-6
[57] pillar_1.9.0 affyio_1.70.0 KernSmooth_2.23-21 generics_0.1.3
[61] vroom_1.6.4 RCurl_1.98-1.12 hms_1.1.3 munsell_0.5.0
[65] scales_1.2.1 gtools_3.9.4 xtable_1.8-4 glue_1.6.2
[69] tools_4.3.1 hexbin_1.28.3 RSpectra_0.16-1 locfit_1.5-9.8
[73] grid_4.3.1 colorspace_2.1-0 GenomeInfoDbData_1.2.10 cli_3.6.1
[77] textshaping_0.3.7 fansi_1.0.4 viridisLite_0.4.2 S4Arrays_1.0.5
[81] gtable_0.3.4 sass_0.4.7 digest_0.6.33 farver_2.1.1
[85] htmltools_0.5.6 lifecycle_1.0.3 mime_0.12 bit64_4.0.5
[89] openssl_2.1.1