
Hi all,
I am using VariantAnnotation::locateVariants to get the location (i.e. intron, intergeneic, ...) of a list of SNPs. I have noticed however, that the results returned by wrong, when I manually look them up in the UCSC genome browser. Here is one such example (I am using R.4.2.2, TxDb.Hsapiens.UCSC.hg19.knownGene 3.2.2 and VariantAnnotation 1.44.1):
library(VariantAnnotation)
library(TxDb.Hsapiens.UCSC.hg19.knownGene)
txdb <- TxDb.Hsapiens.UCSC.hg19.knownGene
snp <- GRanges(seqnames="chr1", ranges = 109706393) # corresponds to SNP rs649539
locateVariants(snp, txdb, AllVariants(), ignore.strand=T)
returns the following result:
GRanges object with 10 ranges and 9 metadata columns:
seqnames ranges strand | LOCATION LOCSTART LOCEND QUERYID TXID
<Rle> <IRanges> <Rle> | <factor> <integer> <integer> <integer> <character>
[1] chr1 109706393 + | intron 49314 49314 1 1785
[2] chr1 109706393 + | intron 49314 49314 1 1786
[3] chr1 109706393 + | intron 135483 135483 1 1786
[4] chr1 109706393 + | intron 49435 49435 1 1787
[5] chr1 109706393 + | intron 49314 49314 1 1788
[6] chr1 109706393 + | intron 49314 49314 1 74189
[7] chr1 109706393 + | intron 49314 49314 1 74190
[8] chr1 109706393 + | intron 135483 135483 1 74190
[9] chr1 109706393 + | intron 49435 49435 1 74191
[10] chr1 109706393 + | intron 135191 135191 1 74191
CDSID GENEID PRECEDEID FOLLOWID
<IntegerList> <character> <CharacterList> <CharacterList>
[1] 5825
[2] 5825
[3] 5825
[4] 5825
[5] 5825
[6] 150365
[7] 150365
[8] 150365
[9] 150365
[10] 150365
-------
seqinfo: 1 sequence from an unspecified genome; no seqlengths
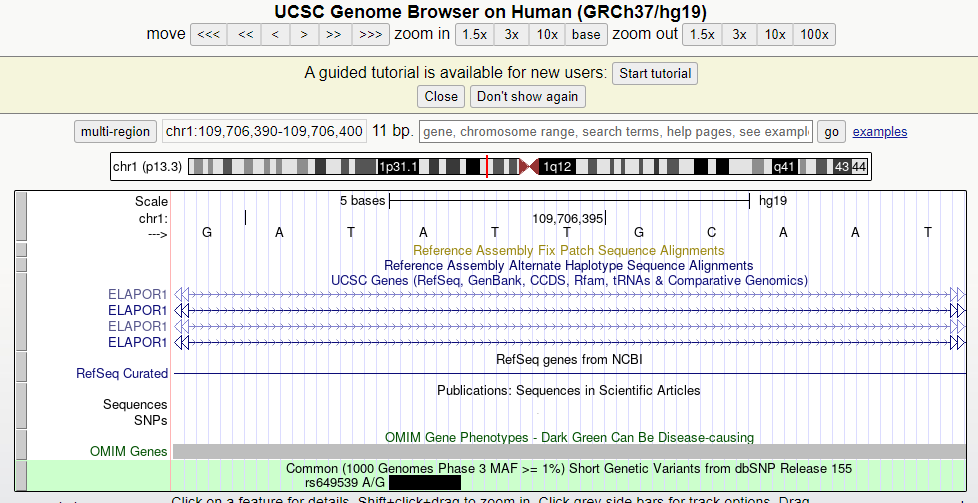
Gene ID 5825 corresponds to the ABCD3 gene and gene id 150365 corresponds to the MEI1 gene. However, if I go to the position of the SNP in the UCSC genome browser, it shows me that I am in an intronic region of ELAPOR1 (see image below).
Furthermore, if I use txdb to look up the chromosome on which these genes are located I get the following results:
select(txdb, keys = c("5825", "150365"), columns="TXCHROM", keytype="GENEID")
leads to:
GENEID TXCHROM
1 5825 chr1
2 150365 chr22
I would very much appreciate any insights into what is happening here, because it seems like the tool is just completely off in this case.

Thank you very much for your quick reply! I guess if I do not know a priori where the SNP is located, I have to iterate thorugh the different
intronsByTranscript,fiveUTRsByTranscript,threeUTRsByTranscript,...? Or is there a way to combine the outputs from all of these commands?You can also just stuff that 'z' object into an env and use that.
Thank you so much! Just for other people that stumble across this, I have tried to replicate the cache that is created by
VaroiantAnnotationlike this.You can certainly do that, but it's extra unnecessary work, as the rest of the cache will already be correctly populated. You only need to do what I showed you.