
Hi, I am trying to do a WGCNA analysis on my bulk RNAseq data.
I have 3 cell lines, and conditions (for each cell line): control, ExposureLow, ExposureHigh; and I am also doing a surrogate variable analysis to consider as non interesting covariates.
i am interested in the Exposure effect.
A1) regarding normalization: I am trying to use voom and calcNormFactors to normalize and prepare the data for WGCNA; and I am also regressing out the effect of Line+ the SVs by calculating this beta coeficient and substracting it from the normalizes counts. I do get a very nice PCA clustering by Exposure condition. But I wonder if this preprocessing of the data is correct.
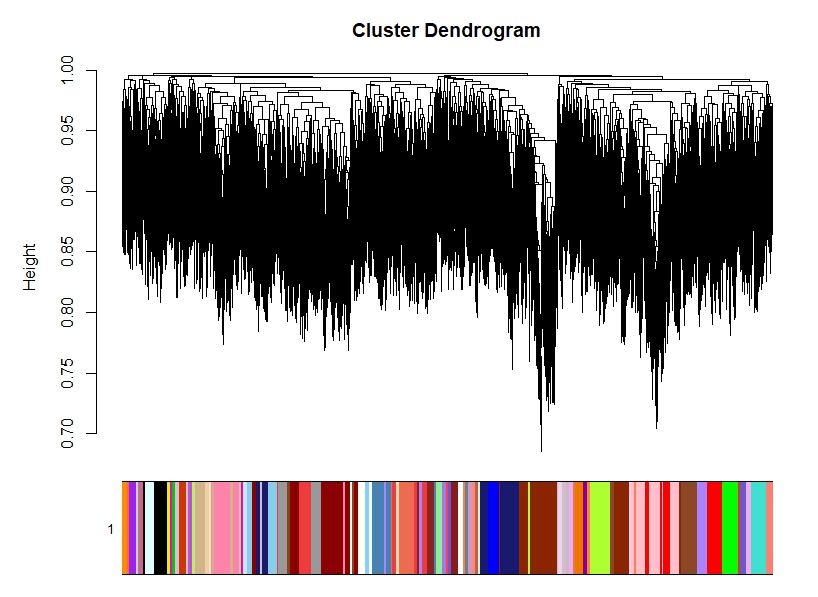
A2) Regarding WGCNA, the free scale topology suggest me a power of 28 for > 0.8< and with blockwiseModules() the tree is awful, very noisy (see image). I can still play with the parameters and get some modules, even significant for my conditions, but is this ok?
B1) I also tried normalization with DEseq2 but the PCA clusters are by line and not by exposure. I wonder if I can correct for the line and svs as with voom... Or if I am missing some other transformation.
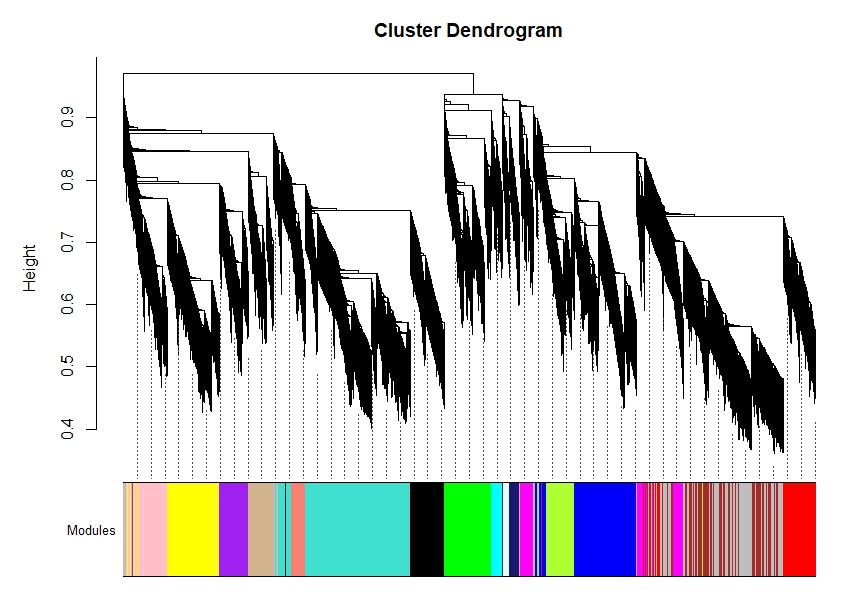
B2) the scale free topology is much better, I could pick a power of 7. and blockwiseModules() are very different and the modules are then no significant for my conditions.
C) I also have TPM already from RSEM... in case is better than starting from read counts, should I still do A or B or just log transform?
Are both approaches correctly done? Which one do you think is best given my data and results?
Why removing genes with low counts in approach B but not in A? Should I nut be using same criteria?
Thank you in advance!
#A1 norm by voom
design=model.matrix(~Exposure + Line +sv1+ sv2+ sv3+sv4+ sv5,
data = GenX_meta)
dge.voom = voom(calcNormFactors(DGEList(counts = counts),
method = 'TMM'), design, plot = T)
Y = as.matrix(dge.voom$E)
X = as.matrix(design)
beta = (solve(t(X)%*%X)%*%t(X))%*%t(Y)
regressed_dataX = Y - t(X[,c(4:ncol(X))] %*% beta[c(4:nrow(beta)),])
##A2
powers = c(seq(1,9,by=1),seq(10,30,by=2))
sft_cor = pickSoftThreshold(data= t(regressed_dataX),
networkType = "signed", corFnc="cor",verbose=2,
powerVector=powers,blockSize = 20000)
plot(sft_cor$fitIndices[,1],
-sign(sft_cor$fitIndices[,3])*sft_cor$fitIndices[,2],
xlab="Soft Thresh Power", ylab="Scale free R^2",type="n")
text(sft_cor$fitIndices[,1],
-sign(sft_cor$fitIndices[,3])*sft_cor$fitIndices[,2],
labels = powers, cex = 0.7, col="red", xlab="Soft Thresh Power",
ylab="Scale free R^2")
abline(h=0.8, col="black")
plot(sft_cor$fitIndices[,1], sft_cor$fitIndices[,5],
xlab = "Soft threshold power", ylab = "Mean connectivity", type = "n")
text(sft_cor$fitIndices[,1], sft_cor$fitIndices[,5],
labels = powers, cex = 0.7, col="black")
net = blockwiseModules(datExpr=t(regressed_dataX), maxBlockSize=15000,networkType="signed",TOMtype="signed", corType="bicor",
power = 28, mergeCutHeight= 0.25, minModuleSize= 40, pamStage=TRUE, reassignThreshold=1e-6,
saveTOMFileBase="WGCNA_signed_cor_counts_regress_28_0.25_5", saveTOMs=TRUE,
verbose = 5, deepSplit=2)
#2A
# create dds
dds <- DESeqDataSetFromMatrix(countData = round(data),
colData = colData,
design = ~ Line + Exposure)
## remove all genes with counts < 10 in more than 75% of samples (16*0.75)
dds75 <- dds[rowSums(counts(dds) >= 10) >= (16*0.75),]
nrow(dds75) # 13525 genes
# perform variance stabilization
dds_norm <- vst(dds75)
# get normalized counts
norm.counts <- assay(dds_norm) %>%
t()
#2B
net = blockwiseModules(datExpr=norm.counts, maxBlockSize=15000,networkType="signed",corType="bicor",
power = 7, mergeCutHeight= 0.25, minModuleSize= 40, pamStage=TRUE, reassignThreshold=1e-6,
saveTOMFileBase="WGCNA_DEseq_signed_cor_counts_regress_7", saveTOMs=TRUE,
verbose = Inf, deepSplit=2)

HI James,
I am sorry, my concerns were actually mainly related to normalization and matrix designs with limma and DEseq2 packages... WGCNA plots just point out to me that I might not be normalizing it correctly (rather than trying to ask about blockwisemodules arguments or issues, I believe the previous steps are the incorrect ones).
thank you for the suggestion on biostar.org. I will try that.
Gimena