Entering edit mode

I found one problem when I tried to download the data:
Error in dplyr::bind_rows():
! Can't combine ..368$Tumor_Seq_Allele2 <character> and ..369$Tumor_Seq_Allele2 <logical>.
Is this a problem in the GDC lib function?
When I use this function to handle the data from TCGA-BLCA and TCGA-SKCM, there is no problem.
But I met this problem when I handle the data from TCGA-BRCA. Could you please provide any suggestion and solution?
The code can seen below:
query <- GDCquery(
project = paste0("TCGA-", cancer_type),
data.category = "Simple Nucleotide Variation",
access = "open",
data.type = "Masked Somatic Mutation",
#workflow.type = "MuSE Variant Aggregation and Masking"
workflow.type = "Aliquot Ensemble Somatic Variant Merging and Masking"
)
GDCdownload(query)
maf <- GDCprepare(query)
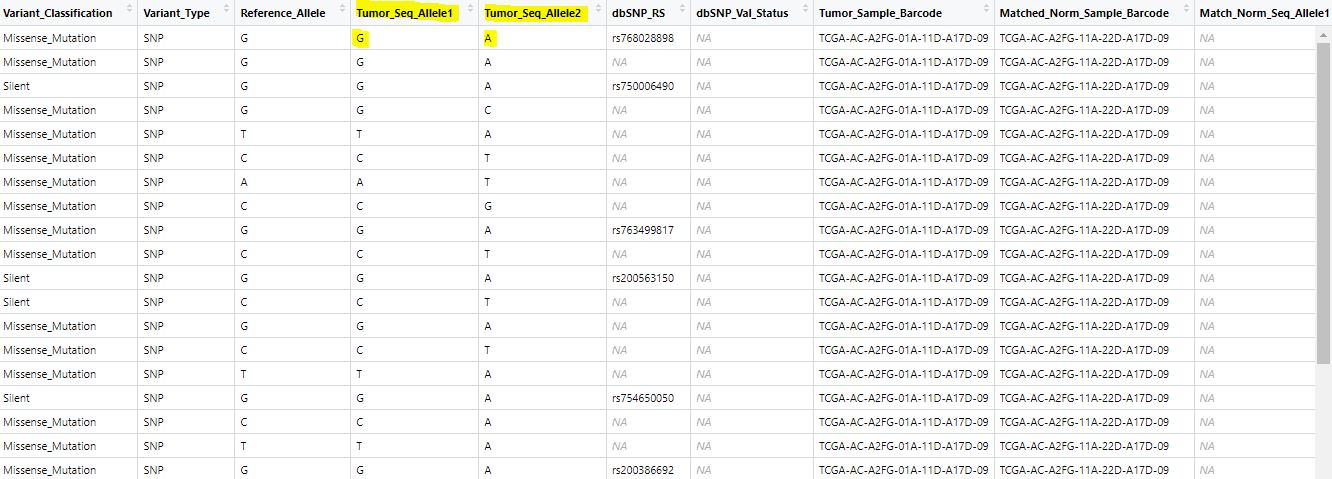
The issue came out when this statement runs: maf <- GDCprepare(query). I checked the data structure:
But I don't know which part includes the combining two 368$Tumor_Seq_Allele2 (character) and ..369$Tumor_Seq_Allele2 (logical).
Hugo_Symbol = col_character(),
Entrez_Gene_Id = col_double(),
Center = col_character(),
NCBI_Build = col_character(),
Chromosome = col_character(),
Start_Position = col_double(),
End_Position = col_double(),
Strand = col_character(),
Variant_Classification = col_character(),
Variant_Type = col_character(),
Reference_Allele = col_character(),
Tumor_Seq_Allele1 = col_character(),
Tumor_Seq_Allele2 = col_character(),
dbSNP_RS = col_character(),
dbSNP_Val_Status = col_logical(),
Tumor_Sample_Barcode = col_character(),
Matched_Norm_Sample_Barcode = col_character(),
Match_Norm_Seq_Allele1 = col_logical(),
Match_Norm_Seq_Allele2 = col_logical(),
Tumor_Validation_Allele1 = col_logical(),
Tumor_Validation_Allele2 = col_logical(),
Match_Norm_Validation_Allele1 = col_logical(),
Match_Norm_Validation_Allele2 = col_logical(),
Verification_Status = col_logical(),
Validation_Status = col_logical(),
Mutation_Status = col_character(),
Sequencing_Phase = col_logical(),
Sequence_Source = col_logical(),
Validation_Method = col_logical(),
Score = col_logical(),
BAM_File = col_logical(),
Sequencer = col_logical(),
Tumor_Sample_UUID = col_character(),
Matched_Norm_Sample_UUID = col_character(),
HGVSc = col_character(),
HGVSp = col_character(),
HGVSp_Short = col_character(),
Transcript_ID = col_character(),
Exon_Number = col_character(),
t_depth = col_double(),
t_ref_count = col_double(),
t_alt_count = col_double(),
n_depth = col_double(),
n_ref_count = col_logical(),
n_alt_count = col_logical(),
all_effects = col_character(),
Allele = col_character(),
Gene = col_character(),
Feature = col_character(),
Feature_type = col_character(),
One_Consequence = col_character(),
Consequence = col_character(),
cDNA_position = col_character(),
CDS_position = col_character(),
Protein_position = col_character(),
Amino_acids = col_character(),
Codons = col_character(),
Existing_variation = col_character(),
DISTANCE = col_logical(),
TRANSCRIPT_STRAND = col_double(),
SYMBOL = col_character(),
SYMBOL_SOURCE = col_character(),
HGNC_ID = col_character(),
BIOTYPE = col_character(),
CANONICAL = col_character(),
CCDS = col_character(),
ENSP = col_character(),
SWISSPROT = col_character(),
TREMBL = col_character(),
UNIPARC = col_character(),
UNIPROT_ISOFORM = col_character(),
RefSeq = col_character(),
MANE = col_character(),
APPRIS = col_character(),
FLAGS = col_logical(),
SIFT = col_character(),
PolyPhen = col_character(),
EXON = col_character(),
INTRON = col_logical(),
DOMAINS = col_character(),
`1000G_AF` = col_double(),
`1000G_AFR_AF` = col_double(),
`1000G_AMR_AF` = col_double(),
`1000G_EAS_AF` = col_double(),
`1000G_EUR_AF` = col_double(),
`1000G_SAS_AF` = col_double(),
ESP_AA_AF = col_double(),
ESP_EA_AF = col_double(),
gnomAD_AF = col_double(),
gnomAD_AFR_AF = col_double(),
gnomAD_AMR_AF = col_double(),
gnomAD_ASJ_AF = col_double(),
gnomAD_EAS_AF = col_double(),
gnomAD_FIN_AF = col_double(),
gnomAD_NFE_AF = col_double(),
gnomAD_OTH_AF = col_double(),
gnomAD_SAS_AF = col_double(),
MAX_AF = col_double(),
MAX_AF_POPS = col_character(),
gnomAD_non_cancer_AF = col_double(),
gnomAD_non_cancer_AFR_AF = col_double(),
gnomAD_non_cancer_AMI_AF = col_double(),
gnomAD_non_cancer_AMR_AF = col_double(),
gnomAD_non_cancer_ASJ_AF = col_double(),
gnomAD_non_cancer_EAS_AF = col_double(),
gnomAD_non_cancer_FIN_AF = col_double(),
gnomAD_non_cancer_MID_AF = col_double(),
gnomAD_non_cancer_NFE_AF = col_double(),
gnomAD_non_cancer_OTH_AF = col_double(),
gnomAD_non_cancer_SAS_AF = col_double(),
gnomAD_non_cancer_MAX_AF_adj = col_double(),
gnomAD_non_cancer_MAX_AF_POPS_adj = col_character(),
CLIN_SIG = col_character(),
SOMATIC = col_character(),
PUBMED = col_logical(),
TRANSCRIPTION_FACTORS = col_logical(),
MOTIF_NAME = col_logical(),
MOTIF_POS = col_logical(),
HIGH_INF_POS = col_logical(),
MOTIF_SCORE_CHANGE = col_logical(),
miRNA = col_logical(),
IMPACT = col_character(),
PICK = col_double(),
VARIANT_CLASS = col_character(),
TSL = col_double(),
HGVS_OFFSET = col_double(),
PHENO = col_character(),
GENE_PHENO = col_double(),
CONTEXT = col_character(),
tumor_bam_uuid = col_character(),
normal_bam_uuid = col_character(),
case_id = col_character(),
GDC_FILTER = col_logical(),
COSMIC = col_character(),
hotspot = col_character(),
RNA_Support = col_character(),
RNA_depth = col_logical(),
RNA_ref_count = col_logical(),
RNA_alt_count = col_logical(),
callers = col_character()