
Hi,
Thanks for your attention.
I am trying to use voomLmFit to compare 3 phenotypes to the same control while also blocking for subject_ID that are repeatedly measured in batch 1 and batch2.
Experimental design
# print meta data
> pb.colData
condition subject_ID_unique subject_ID batch
01501_b2 0_1 01501_b2 01501 b2
04901_b2 0_0 04901_b2 04901 b2
21201_b2 1_1 21201_b2 21201 b2
31101_b1 1_0 31101_b1 31101 b1
31101_b2 1_0 31101_b2 31101 b2
44501_b1 0_0 44501_b1 44501 b2
45401_b2 0_0 45401_b2 45401 b2
51301_b1 0_0 51301_b1 51301 b1
51801_b1 1_1 51801_b1 51801 b1
51801_b2 1_1 51801_b2 51801 b2
51901_b1 0_1 51901_b1 51901 b1
51901_b2 0_1 51901_b2 51901 b2
52001_b2 0_0 52001_b2 52001 b2
53201_b1 0_1 53201_b1 53201 b1
53201_b2 0_1 53201_b2 53201 b2
57001_b2 0_0 57001_b2 57001 b2
61101_b1 0_0 61101_b1 61101 b1
62001_b1 1_0 62001_b1 62001 b1
65301_b2 0_0 65301_b2 65301 b2
71301_b2 0_0 71301_b2 71301 b2
71501_b2 0_0 71501_b2 71501 b2
73801_b2 0_0 73801_b2 73801 b2
77601_b1 1_0 77601_b1 77601 b1
77601_b2 1_0 77601_b2 77601 b2
81001_b2 0_0 81001_b2 81001 b2
87501_b1 1_1 87501_b1 87501 b1
87501_b2 1_1 87501_b2 87501 b2
89201_b1 0_0 89201_b1 89201 b2
A9501_b2 0_0 A9501_b2 A9501 b2
The above table is my meta data where condition has four unique elements, with 0_0 being control and others being diseases. subject_ID is the identifier for every sample. batch is a factor I wish to regress out. subject_ID_unique is subject_ID plus batch to make a unique identifier.
Design matrix and code for running comparison
# condition & batch are factors with set levels
group <- pb.colData$condition
subject_ID <- pb.colData$subject_ID
batch <- pb.colData$batch
design <- model.matrix(~0+group+batch)
# dpb is a DGEList
v <- voomLmFit(counts = dpb,
design = design,
block = subject_ID,
sample.weights = TRUE)
# example contrast
contrast <- makeContrasts(group1_1 - group0_0, levels=colnames(design))
fit <- contrasts.fit(v, contrasts = contrast)
fit <- eBayes(fit)
If the above code makes sense to you (please correct, if not), by comparing 1_1 with 0_0, there were over 900 differentially expressed genes. However, it concerned me because I only have 3 unique samples in 1_1 group and the signals seem driven by outliers.
# print n unique samples per condition
> print(pb.colData %>%
+ distinct(subject_ID, .keep_all = TRUE) %>% select(condition) %>% table())
condition
0_0 0_1 1_0 1_1
14 3 3 3
Here is the PCA for all samples where you can see the major variance is driven by outliers.
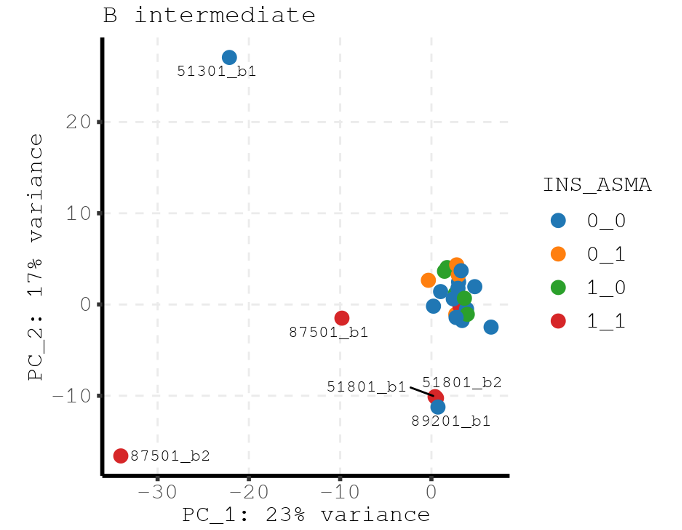
Question 1
How can I get a relatively robust result given the small sample size and outliers? I noticed eBayes has a robust argument. Can I use it? Is there any other methods worth trying?
========================
I am also trying to get the covariance matrix (diagonal and off diagonal) for using as an input for mashr. I read from the limma tutorial section 13.2:
The covariance matrix of the estimated Bhat is Sigma^2 CT (XT Vg X) -1C
Sorry about the format. The formula is on page 62
https://www.bioconductor.org/packages/devel/bioc/vignettes/limma/inst/doc/usersguide.pdf
Question 2
I understand X and C are design matrix and contrast matrix, but what is Vg? How to derive Vg?
Thank you very much for your time and help.
Jiacheng Ding

Thank you very much for your rapid response. Your answer makes a lot sense to me. I check the dimension reduction plot again using
limma::plotMDSwith the following code:It's indeed different from the original PCA plot I provided.
==========
Thanks for your explanation on Vg, however I still don't understand how to get the block correlation. Could you please provide more details on instructing me to calculate it? Thank you very much for your time!
Jiacheng Ding
I have edited my answer above to add code for Vg.
Thank you very very much for your time, professor!
Jiacheng
Your MDS plot is better than before, but you are still not following the recommendation for making for making such plots given in the limma and edgeR documentation. If
dpbis matrix of counts, then we recommend that you use:If
dpbis a DGEList object, then you could usedirectly, which will give the same result. We don't recommend that you use
plotMDS()on thevoom()output becausevoom()output is only intended to be used in functions that can use weights andplotMDS()doesn't use the weights.By the way, you never need to run
voom()aftervoomLmFit()because the output is already stored in thev$EListcomponent of thevoomLmFit()output. You could find that out by reading thevoomLmFithelp page.Thanks a lot for your answer again!
I am trying to validate the solution you provided by comparing the
sqrt(diag(CT (XT Vg X) -1 C))withfit$stdev.unscaled. Based on limma tutorial, they are supposed to be equal.https://www.bioconductor.org/packages/devel/bioc/vignettes/limma/inst/doc/usersguide.pdf page 62
R Code for calculating first gene's stdev.unscaled
My question
I think my way for getting 'weights' is wrong considering
stdev.unscaled.calculateddoes not equal tofit$stdev.unscaled[1,]. I understand I can usefit$stdev.unscaled[1,]to back calculateweightswith your solution, however, how to correctly get theweightsforv?Thank you very much!
Jiacheng Ding
The weights are contained in
v$EList$weights. I previously mistyped it asv$weightsbut have now corrected the code in my answer above.Here is an reproducible example of computing the covariance matrix:
In the above example, the covariance matrix agrees exactly with
fit$stdev.unscaled. Beware though that, if you usecontrasts.fit, then you will not be able to reproducefit$stdev.unscaledexactly for the reason explained on thecontrasts.fithelp page.Thank you very much, professor! It worked for my data.
Thank you for your time. Jiacheng