
Hi,
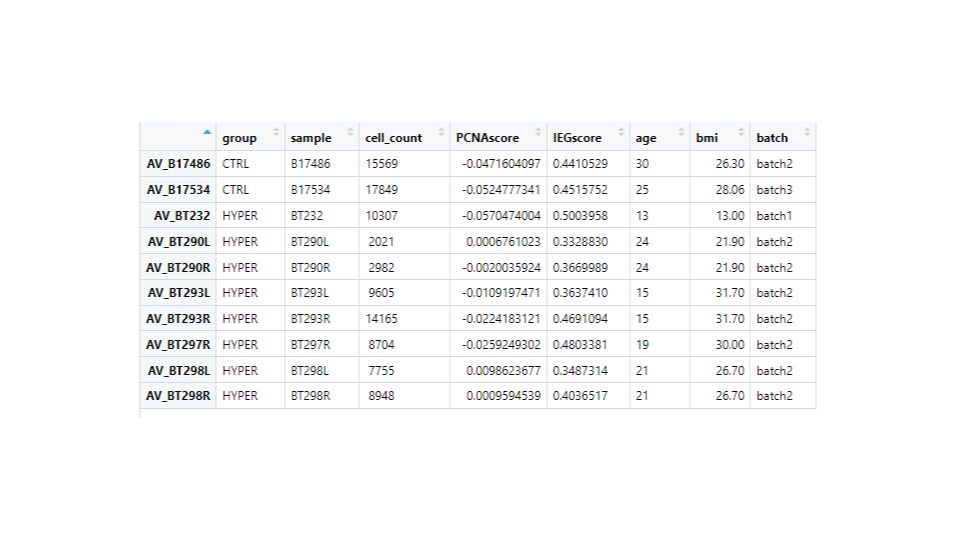
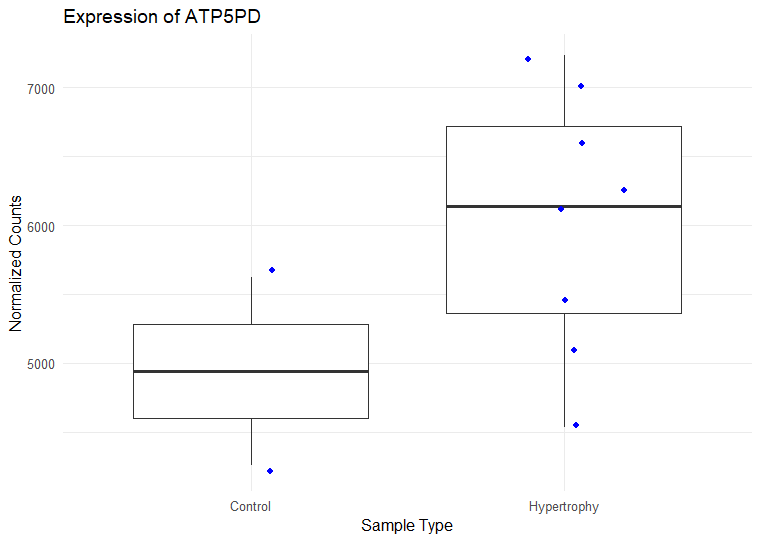
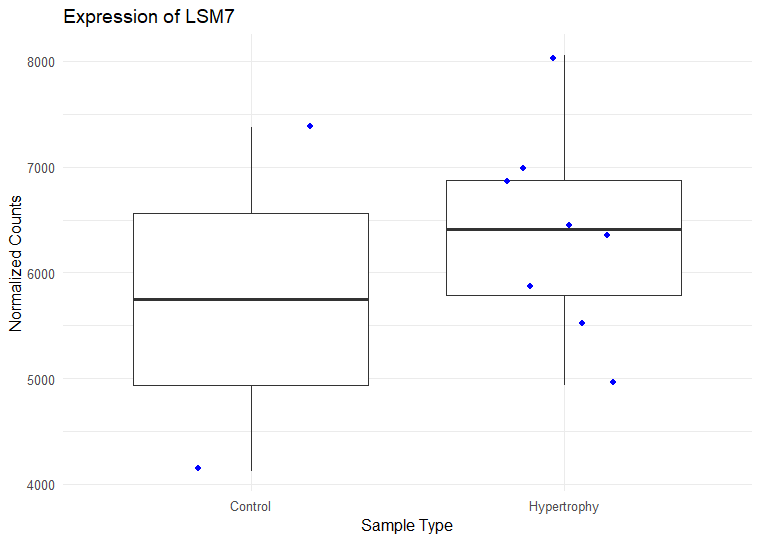
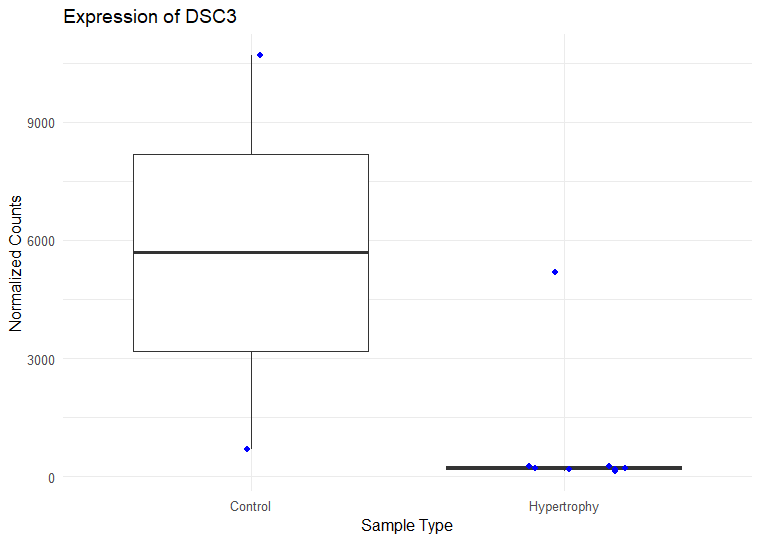
I have ran DESeq2 to get log2 fold changes of hypertrophy samples relative to control. (Added my code below). I think I am setting my contrast correct of Hypertrophy vs. control correctly while getting results. My log2 fold change results are mostly in concordance with what I observe in normalized counts. Meaning, for a gene, if the control samples have a lower mean than the hypertrophy samples, they show a positive log fold change and vice versa However, for some genes the results are conflicting. For example, The gene ATP5D is shown to have a negative log2 fold change of -0.48 but when I look at the normalized counts of the gene, the gene clearly has higher mean in the hypertrophy samples than the control (shown below). Or LSM7 has a negative log2 fold change of -0.5 while the normalized counts of Hypertrophy samples show a higher mean. On the other hand DSC3 has a negative log2 fold change of -2.13 and that can be observed in the normalized counts (shown below). Would appreciate any insight into why this might occur. I have also included a picture of my metadata.
# Create DESeq2 object
dds <- DESeqDataSetFromMatrix(counts,
colData = md,
design = ~PCNAscore + batch + group)
# Run DESeq2 differential expression analysis
dds <- DESeq(dds)
# Check the coefficients for the comparison
resultsNames(dds)
# Generate results object
#res <- results(dds)
res <- results(dds, contrast=c("group","HYPER","CTRL"), alpha = 0.05)
[1] "Intercept" "PCNAscore" "batch_batch2_vs_batch1" "batch_batch3_vs_batch1"
[5] "group_HYPER_vs_CTRL"
# Shrink the log2 fold changes to be more appropriate using the apeglm method - should cite [paper]() when using this method
res <- lfcShrink(dds,
coef = "group_HYPER_vs_CTRL",
res=res,
type = "apeglm")
res_tbl <- res %>%
data.frame() %>%
rownames_to_column(var = "gene") %>%
as_tibble() %>%
arrange(padj)
sessionInfo( )

That makes sense. I am running iterations without any confounders, and with confounders to see how much of an effect each has. Appreciate your help.