
Hello,
I am running into a weird problem where I:
- Filter out non-primary alignments of a bam file, as well as some other things (
samtools view -@ 8 -b -h -q 2 -F 0x4 -F 0x8 -F 0x100 -F 0x200 -F 0x400 -F 0x800 WT_1.bam > WT_1.f.bam) - Assign reads to genes with featureCounts (call below).
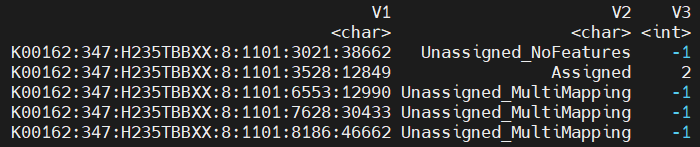
Since only the primary alignment of multi-mapping reads are retained, there should be no instances of failed feature assignments due to the reason of "Unassigned_MultiMapping". Thus, I was surprised to find that around 40% of the reads in this and several other bam file were flagged as "Unassigned_MultiMapping" in the resulting CORE file.
featureCounts call:
featureCounts -T 8 -s 2 -a genome.gtf --primary -R CORE -g gene_id -t transcript -p --countReadPairs -o WT_1.featureCounts bams/WT_1.f.bam
The same thing happens with these other calls, such as assignment of reads to exclusively exonic regions:
featureCounts -T 8 -s 2 -a genome.gtf --primary --nonOverlap 0 -R CORE -g gene_id -J -p --countReadPairs -o WT_1.featureCounts bams/WT_1.f.bam
or with a specialized custom annotation and an attempt to assign reads to regions near exon-exon junctions:
featureCounts -T 8 -s 2 -a junctions.gtf --primary -R CORE -g junction_id -t eej -O --fracOverlapFeature 0.9 -p --countReadPairs -o WT_1.featureCounts bams/WT_1.f.bam
In all cases, all of the exact same reads get flagged as "Unassigned_MultiMapping". These reads are all successfully assigned if I replace --primary with -M in my featureCounts call. This behavior seems to contradict the documentation for the --primary flag, which reads "All primary alignments in a dataset will be counted no matter they are from multimapping reads or not (i.e. -M is ignored)."
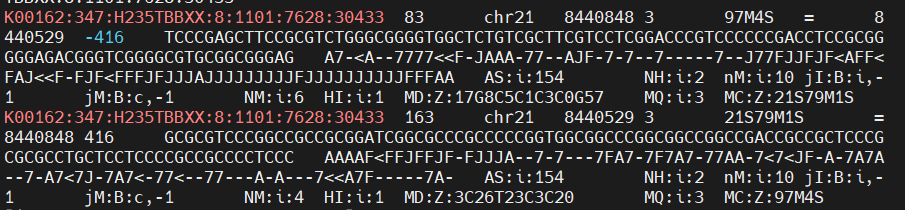
I have attached a screenshot of data for one of the reads that gets unassigned due to being multi-mapping, to show that the non-primary flag is not on. All of the bam files and standard annotation that I used to first identify this problem are in the .test/ folder of this repo.
CORE file snippet:
One unassigned read:
I am happy to provide any additional information,
Isaac