
Hi, I'm trying to do two-factor DEG analysis using DESeq2.
colData(dds)
condition batch infection metabolite sizeFactor
<factor> <factor> <factor> <factor> <numeric>
Mock1 Mock first Not-Infected Non 0.904435
Mock2 Mock first Not-Infected Non 1.060912
Mock3 Mock second Not-Infected Non 0.864510
Mock4 Mock third Not-Infected Non 1.064498
L1 L third Not-Infected L 1.137483
... ... ... ... ... ...
V4 Virus third Infected Non 1.104777
LV1 L+Virus first Infected L 0.862166
LV2 L+Virus first Infected L 0.897168
LV4 L+Virus third Infected L 1.142766
LV5 L+Virus third Infected L 1.160191
What really matters are infection and metabolite columns and I ran the following code.
dds_fin<-DESeqDataSetFromMatrix(countdata_fin,colData = metadata_fin,design = ~ infection + metabolite + infection:metabolite)
To analyze the main effect of infection, which means the effect of infection in non-treated group, I ran the following code.
res_infection<- results(dds_fin,contrast = c("infection","Infected","Not-Infected"))
The results said there is only one gene (GNAT1) that are differentially expressed with statistical significance and high log2FoldChange value (between mock and V group as far as I understand correctly). My question is, if I see the normalized expression of GNAT1 gene like the image I posted, there is almost no difference between Mock and Virus-infected(V) group. I'm really confused with this result.
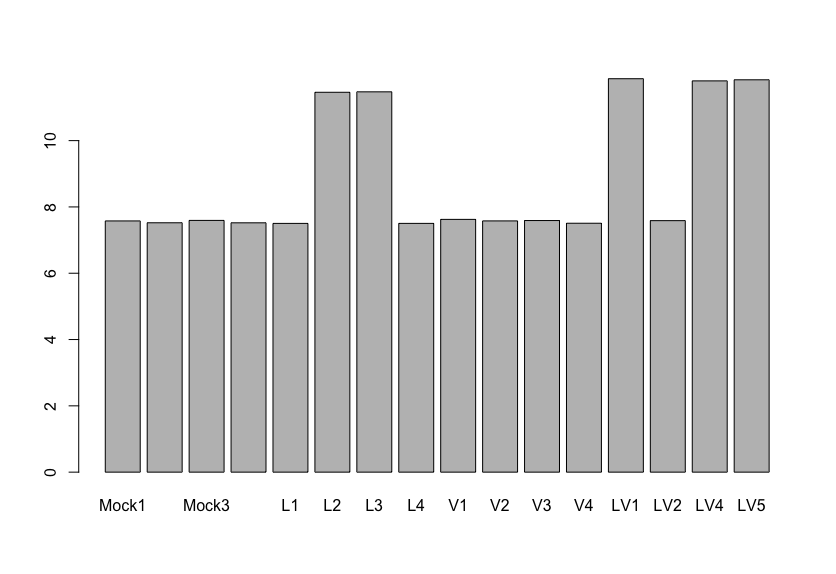
And another question is about contrast. What I really want to know is the effect of infection regardless of what is treated (control or metabolite L). I saw deseq2: coding 2x2 design , where situation seems similar. So if I want to extract the pure infection effect, do I need to do numerical contrast?
I appreciate any guidance or advice. Thanks a lot!
JY
